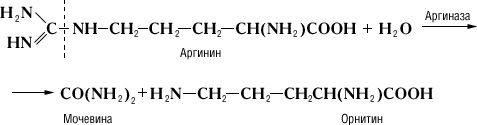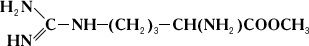Ферменти (від латів.(латинський) fermentum – закваска), ензими, специфічні білкові каталізатори, присутні у всіх живих клітинах. Майже всі біохімічні реакції, що протікають в будь-якому організмі і в своєму закономірному поєднанні складові його обмін речовин, каталізують відповідними Ф. Направляя і регулюючи обмін речовин, Ф. грають найважливішу роль у всіх процесах життєдіяльності.
Як всякі каталізатори, Ф. знижують енергію активації, необхідну для здійснення тієї або іншої хімічної реакції, направляючи її обходним дорогою – через проміжні реакції, які вимагають значно меншої енергії активації. Так, реакція АБ ® А + Б у присутності Ф. йде таким чином: АБ + Ф ® АБФ і далі АБФ ® БФ + А і БФ ® Би + Ф. Наприклад, для здійснення реакції гідролізу дісахаріда сахарози, в результаті якого утворюються глюкоза і фруктоза, без участі каталізатора потрібний 32 000 кал (1 кал = 4,19 дж ) на міль сахарози. Якщо ж реакція каталізує Ф. b-фруктофуранозідазой, то необхідна енергія активації складає всього 9400 кал. Подібне пониження енергії активації під впливом Ф. – слідство перерозподіли електронної щільності і деякої деформації молекул субстрата, що відбувається при утворенні проміжного з'єднання, – фермент-субстратного комплексу (АБФ). Ця деформація, ослабляючи внутрішньомолекулярні зв'язки, приводить до пониження необхідної енергії активації і, отже, прискорює перебіг реакції (див. Каталіз, Ферментативний каталіз ) .
Історія вивчення ферментів. В 1814 русявий.(російський) хімік До. Р. С. Кирхгоф відкрив ферментативну дію водних витягів з пророслого ячменю, що розщеплювали крохмаль до цукру. Можна вважати, що ці роботи поклали початок ензімологиі (ферментології) як самостійному розділу біологічної хімії. У 1833 французькими хіміками А. Пайеном і Ж. Персо вперше був виділений з солоду препарат ферменту амілази, що сприяло розвитку препаратівной хімії Ф. У середини 19 ст розгорілася дискусія про природу бродіння між Л. Пастером, з одного боку, і Ю. Лібіхом, П. Е. М. Бертло і До. Бернаром – з іншою. Спираючись на свої класичні роботи, Пастер розвивав уявлення про те, що бродіння викликається лише живими мікроорганізмами і що процес бродіння нерозривно пов'язаний з їх життєдіяльністю. Лібіх і його прибічники, відстоюючи хімічну природу бродіння, вважали, що воно є наслідком освіти в клітках мікроорганізмів розчинних Ф., подібних до амілази, що виділяється з солоду. Проте всі спроби виділити із зруйнованих дріжджових кліток розчинний Ф., здатний викликати бродіння, не удавалися. Дискусія Лібіха і Пастера про природу бродіння була дозволена в 1897 Е. Бухнером, який, розтираючи дріжджі із землею інфузора, виділив з них безклітинний розчинний ферментний препарат (названий їм зимазою), що викликав спиртне бродіння. Відкриття Бухнера затвердило матеріалістичне розуміння природи бродінь і мало велике значення для подальшого розвитку як ензімологиі, так і всієї біохімії.
На початку 20 ст Р. Вільштеттер із співробітниками став широко застосовувати для виділення і очищення Ф. метод адсорбції (вперше запропонований А. Я. Данільовським для розділення Ф. підшлункової залози). Роботи Вільштеттера, що мали велике значення для характеристики властивостей окремих Ф., привели в той же час до принципово неправильного виводу, що Ф. не належать ні до одного з відомих класів органічних сполук. Видатним успіхом в з'ясуванні хімічної природи Ф. були дослідження американських біохіміків Дж. Самнера, що виділив в 1926 в кристалічному вигляді Ф. уреазу з насіння канавалії, і Дж. Нортропа, що отримав в 1930 кристалів протеолітичного Ф. пепсину. Роботи Самнера і Нортропа вказали дорогу здобуття високоочищених кристалічних препаратів Ф. і в той же час неспростовно довели білкову природу Ф.
З середині 20 ст завдяки розвитку методів физико-хімічного аналізу (головним чином хроматографії ) і методів білкової хімії розшифрована первинна структура багатьох Ф. Так, роботами американських біохіміків С. Мура, У. Стайна і К. Анфінсена показане, що Ф. рібонуклеаза з підшлункової залози бика є поліпептидним ланцюжком, що складається з 124 амінокислотних залишків, сполучених в 4 місцях дисульфідними зв'язками.
За допомогою рентгеноструктурного аналізу розшифрована вторинна і третинна структура ряду Ф. Так, методом рентгеноструктурного аналізу англійський учений Д. Філліпс в 1965 встановив тривимірну структуру Ф. лізоциму . Показане, що багато Ф. володіють також четвертинною структурою, тобто їх молекула складається з декількох ідентичних або різних по складу і структурі білкових субодиниць (див. Біополімери ) .
Загальна характеристика ферментів. Все Ф. розділяються на дві великі групи: однокомпонентні, такі, що складаються виключно з білка, і двокомпонентні, такі, що складаються з білка, званого апоферментом, і небілкової частини, званою простетічеськой групою. Апофермент двокомпонентних Ф. називають також білковим носієм, а простетічеськую групу – активною групою. Завдяки роботам О. Варбурга, А. Теорелля, Ф. Лінена, Ф. Ліпмана і Л. Лелуара встановлене, що простетічеськие групи багатьох Ф. є похідними вітамінів або нуклеотидів . Т. о. був відкритий найважливіший функціональний зв'язок між Ф., вітамінами і нуклеотидами, будівельною «цеглою» нуклеїнових кислот, що є.
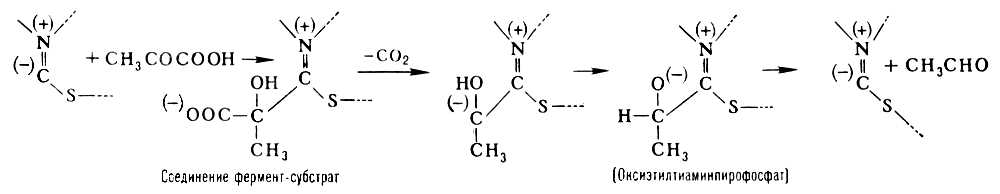
Прикладом двокомпонентного Ф. є піруватдекарбоксилаза, що каталізує розщеплювання піровиноградної кислоти на двоокис вуглецю і оцетовий альдегід: Ch 3 COCOOH ® Ch 3 CHO + Co 2 . Простетічеськая група піруватдекарбоксилази (тіаміннірофосфат) утворена молекулою тіаміну (вітаміну B 1 ) і двома залишками фосфорної кислоти. Простетічеськие групи ряду важливих окислювально-відновних Ф. – дегідрогенази містять похідне аміда нікотинової кислоти (Ніацину), або ж рибофлавіну (вітаміну B 2 ); до складу простетічеських групи т.з. пірідоксальових ферментів, аміногруп (–NH 2 ), що каталізують перенесення, і декарбоксилювання і ряд ін. перетворень амінокислот, входить пірідоксальфосфат – похідне вітаміну B 6 ; активна група Ф., залишків різних органічних кислот, що каталізують перенесення (наприклад, ацетилу Ch 3 CO–), включає вітамін пантотенову кислоту . До двокомпонентних Ф. відносяться також важливі окислювальні Ф. – каталаза (каталізує реакцію розкладання перекису водню на воду і кисень) і пероксидаза (окислює перекисами різні з'єднання, наприклад поліфеноли з освітою відповідного хінону і води). Каталітична дія цих Ф. може бути відтворено за допомогою іонів тривалентного заліза. Ці іони володіють, проте, дуже малою каталітичною активністю, яка може бути посилена, якщо атом заліза входить до складу гема . Хоча гем володіє вже значною каталазним дією, його каталітична активність все ж в декілька мільйонів разів менше активності каталази, в якій гем як простетічеськой група цього Ф. пов'язаний із специфічеськ ним білком. Гем володіє також слабкою пероксидазною дією, проте ця дія виявляється повною мірою лише після з'єднання гема із специфічним білком в цілісний Ф. пероксидазу. Т. о., з'єднання простетічеськой групи з білком приводить до різкого зростання її каталітичної активності. В той же час від природи білка залежить не лише каталітична активність, але і специфічність дії Ф. Міцність зв'язку простетічеськой групи і апофермента різна в різних Ф. В деяких Ф., наприклад в дегідрогенази, що каталізує окислення різних субстратів шляхом відщеплення водню, цей зв'язок є неміцним. Такі Ф. легко диссоціюють (наприклад, при діалізі ) і розпадаються на простетічеськую групу і апофермент. Простетічеськие групи, Ф, що легко відділяються від білкової частини., називаються коферментами .
Багато Ф. містять метали, без яких Ф. не активний. Ці метали називаються кофакторам і. Так, пероксидаза і каталаза містять залізо, аськорбінатоксидаза, що каталізує окислення аскорбінової кислоти, – мідь, алкогольдегидрогеназа, що окислює спирти у відповідні альдегіди, – цинк.
Специфічність і механізм дії ферментів. Дія Ф., на відміну від неорганічних каталізаторів, строго специфічно і залежить від будови субстрата, на який Ф. діє. Прекрасним прикладом такої залежності служить реакція гідролітичного розщеплювання амінокислоти аргініну, що каталізує аргіназою, на орнітин і сечовину:
Проте аргіназа не розщеплює метилового ефіру аргініну:
Дипептид, що складається із залишків двох молекул аргініну, під дією аргінази дає лише половину теоретичної кількості сечовини. Очевидно, що, хоча розщеплювання аргініну відбувається в місці, вельми віддаленому від карбоксильної (COOH) групи (показано пунктиром), необхідною умовою дії аргінази є її з'єднання з карбоксильною групою аргініну. Тому заміщення водню в карбоксильній групі на метільний залишок або ж скріплення карбоксильної групи з другою молекулою аргініну роблять різкий вплив на дію аргінази. Приклади специфічності дії Ф. можуть бути приведені при розгляді їх стереохимічеськой специфічності, тобто дії Ф. на стереоізомери (див. Ізомерія ) . Так, Ф., що окислює природні l-амінокислоті, не діє на d-ізомері цих же амінокислот; Ф. дипептидаза, гидролізірующий діпептіди, що складаються із залишків l-амінокислот, не діє на такі ж діпептіди, що складаються із залишків d-амінокислот. Специфічність дії Ф. послужила йому.(німецький) ученому Е. Фішеру підставою для порівняння субстрата і Ф., який каталізує його перетворення, із замком і відповідним йому ключем. Стереохимічеськая специфічність Ф. найтіснішим чином пов'язана з однією з основних особливостей живих організмів – їх здібністю до синтезу оптично активних органічних сполук.
В утворенні з'єднання між ферментом і субстратом – т.з. фермент-субстратного комплексу – беруть участь лише деякі функціональні групи молекули Ф., створюючі його активний центр . Так, наприклад, в молекулі гидролізірующего білки хімотрипсину, що складається з 246 амінокислотних залишків, активний центр утворений одним із залишків серину (хімотрипсин відноситься до серіновим протєїназам) і двома залишками гістидину, розташованими у віддалених один від одного ділянках поліпептидного ланцюга. Зближення цих функціональних груп активного центру відбувається завдяки властивій молекулі хімотрипсину специфічній просторовій (третинною) структурі. Її порушення в результаті денатурацію білка або яких-небудь хімічних модифікацій приводить до зміни або повної втрати каталітичної активності. В разі двокомпонентних Ф. у утворенні фермент-субстратного комплексу беруть участь не лише функціональні групи апофермента, але і простетічеськая група. Так, при розщеплюванні піровиноградної кислоти піруватдекарбоксилазой субстрат зв'язується з частиною молекули тіамін-пірофосфата таким чином:
Виключно висока специфічність дії Ф. пояснюється їх білковою природою. Так, пірідоксальовиє Ф., що містять один і той же кофермент (пірідоксальфосфат), можуть належати до різних класів і каталізувати найрізноманітніші реакції. Специфічність їх дії залежить від природи апофермента.
Умови дії ферментів. Дія Ф. залежить від ряду чинників, перш за все від температури і реакції середовища (ph). Оптимальна температура, при якій активність Ф. найбільш висока, знаходиться зазвичай в межах 40–50 °С. При нижчих температурах швидкість ферментативної реакції, як правило, знижується, а при температурах, близьких до 0 °С, практично реакція повністю припиняється. При підвищенні температури вище оптимальною швидкість ферментативної реакції також знижується і, нарешті, повністю припиняється. Зниження інтенсивності дії Ф. при підвищенні температури понад оптимальну пояснюється руйнуванням (денатурацією), що головним чином починається, що входить до складу Ф. білка. Оскільки білки в сухому стані денатуруються значно повільніше, ніж білки оводненниє (у вигляді білкового гелю або розчину), інактівірованіє Ф. у сухому стані відбувається набагато повільніше, ніж у присутності вологи. Тому сухі спори бактерій або сухе насіння можуть витримати нагрівання набагато вищих температур, ніж ті ж спори або насіння в зволоженому стані.
Найважливішим чинником, від якого залежить дія Ф., як встановив вперше С. Серенсен, є активна реакція середовища – ph. Окремі Ф. розрізняються по оптимальній для їх дії величині ph. Так, наприклад, пепсин, що міститься в шлунковому соку, найбільш активний в сильнокислой середовищі (ph 1–2); трипсин – протеолітичний Ф., що виділяється підшлунковою залозою, має оптимум дії в слаболужному середовищі (ph 8–9); оптимум дії папаїна – протеолітичного Ф. рослинного походження – знаходиться в слабокислому середовищі (ph 5–6).
Дія Ф. залежить також від присутності специфічних активаторів і неспецифічних або специфічних інгібіторів. Так, ентерокіназа, що виділяється підшлунковою залозою, перетворює неактивний трипсиноген на активний трипсин. Подібні неактивні Ф., що містяться в клітках і в секретах різних залоз, називаються проферментамі . Багато Ф. активуються у присутності з'єднань, що містять сульфгідрильну групу (–sh). До них належать амінокислота цистеїн і трипептид глутатіон, що міститься в кожній живій клітині. Особливо сильна активуюча дія глутатіон надає на деяких протеолітичних і окислювальні Ф. Неспецифічеськоє пригноблення (інгібірування) Ф. відбувається під дією різних речовин, що дають з білками нерозчинні осідання або блокуючих в них які-небудь групи (наприклад, sh-групі). Існують більш специфічні інгібітори Ф., пригноблення якими каталітичних функцій засновано на специфічному пов'язанні цих інгібіторів з певними хімічними угрупуваннями в активному центрі Ф. Так, окисел вуглецю (CO) специфічно інгібірує ряд окислювальних Ф., що містять в активному центрі залізо або мідь. Вступаючи в хімічну сполуку з цими металами, вона блокує активний центр Ф. і внаслідок цього він втрачає свою активність. Розрізняють оборотне і необоротне інгібірування Ф. В разі оборотного інгібірування (наприклад, дія малонової кислоти на сукцинатдегидрогеназу) активність Ф. відновлюється при видаленні інгібітору діалізом або іншим способом. При необоротному інгібіруванні дія інгібітору, навіть при дуже низьких його концентраціях, посилюється з часом і врешті-решт настає повне гальмування активності Ф. Інгибірованіє Ф. може бути конкурентним і неконкурентним. При конкурентному інгібіруванні інгібітор і субстрат конкурують між собою, прагнучи витіснити один іншого з фермент-субстратного комплексу. Дія конкурентного інгібітору знімається високими концентраціями субстрата, тоді як дія неконкурентного інгібітору в цих умовах зберігається. Дія на Ф. специфічних активаторів і інгібіторів має велике значення для регулювання ферментативних процесів в організмі.
Класифікація і номенклатура ферментів. По рекомендації Міжнародного біохімічного союзу, Ф. розділяють на 6 класів: 1) оксидоредуктаза, 2) трансферази, 3) гідролази, 4) ліази, 5) ізомерази, 6) лігази. Рекомендована наступна нумерація Ф. Шифр (індекс) кожного Ф. містить 4 числа, розділених крапками. Перша цифра вказує клас, друга – підклас, третя – подподкласс, четверта, – порядковий номер в даному подподклассе. Так, Ф. аргіназа, що розщеплює аргінін на орнітин і сечовину, має шифр 3.5.3.1, тобто відноситься до класу гидролаз, підкласу Ф., що діють на непептидні С–n-cвязі, і подподклассу Ф., що розщеплюють ці зв'язки в лінійних (не циклічних) з'єднаннях.
Клас оксидоредуктази включає Ф., каталізуючі окислювально-відновні реакції, і розділяється на 14 підкласів залежно від природи тієї групи в молекулі субстрата, яка піддається окисленню (спиртна, альдегідна, кетонна і т.д.). Подподкласси оксидоредуктази індексуються залежно від типа водню (електронів), що бере участь в реакції акцептора, – кофермента, цитохрому, молекулярного кисню і т.д. Т. о., перші три цифри шифру визначають типа Ф., так, наприклад, 1.2.3 позначають оксидоредуктазу, що діє на альдегід з молекулярним киснем як акцептор електронів. Клас трансфераз, об'єднуючий Ф., каталізуючі реакції перенесення груп, підрозділяється на 8 підкласів залежно від природи переносимих груп, якими можуть бути одинвуглецеві або глікозільниє залишки, що азотисті або містять сірку групи і т.д. В трансфераз третя цифра характеризує типа переносимих груп (наприклад, одинвуглецева група може бути метилом, карбоксилом, формілом і т.д.). До гидролазам належать Ф., що каталізують гідролітичне розщеплювання різних з'єднань; розділяються на 9 підкласів залежно від типа гідролізованого зв'язку – сложноефірной, пептидною, глікозідной і т.д. Третя цифра в гидролаз уточнює тип гідролізованого зв'язку. Ліази – Ф., що відщеплюють від субстрата ту або іншу групу (негідролітичними дорогами) з утворенням подвійного зв'язку або, навпаки, що приєднують групи до подвійних зв'язків. В ліаз 5 підкласів, друга цифра шифру позначає типа такою, що піддається розриву зв'язку (вуглець – вуглець, вуглець – кисень і т.д.), а третя – тип групи, що відщеплюється. Ізомерази, що каталізують реакції ізомеризації, розділяються на 5 підкласів залежно від типа реакції, що каталізує; третя цифра шифру деталізує характер перетворення субстрата. Лігазамі (або синтетазамі) називаються Ф., які каталізують з'єднання двох молекул, зв'язане з розщеплюванням пірофосфатной зв'язку в молекулі аденозинтрифосфорної кислоти (АТФ) або аналогічного тріфосфата. Перша цифра шифру лігаз позначає типа знов утворюваного зв'язку (вуглець – азот, вуглець – кисень і т.д.), а друга – природу з'єднання, що утворюється.
Класифікація і номенклатура Ф., окрім шифру, включає також систематичні і тривіальні (робітники) назви. Так, наприклад, систематична назва карбоксилаза 2-оксокислот відповідає вже згадуваному тривіальному назва піруватдекарбоксилаза, а систематична назва L -apгинін – амідіногидролаза – робітникові назва аргіназа.
Регуляція ферментативних процесів. Дія Ф. у організмі здійснюється шляхом регуляції їх синтезу і активності. Властивий даному організму набір Ф. визначається його генетичною природою. Проте він може змінюватися під впливом різних внутрішніх і зовнішніх чинників – мутацій, дії іонізуючої радіації, складу газового середовища, умов живлення і т.д. Так, в результаті мутацій виникають т.з. «молекулярні хвороби» (наприклад, алкаптонурія). При цьому спадковому захворюванні у хворих з сечею виділяється гомогентізіновая кислота, що утворюється в результаті перетворень амінокислоти Тирозину . Гомогентізіновая кислота накопичується в організмі і виділяється з сечею унаслідок того, що у хворих алкаптонурією загублена здібність до синтезу два Ф., що каталізують її подальше окислення, – параоксифенілпіруватоксидази і оксидаза гомогентізінової кислоти. Вплив умов живлення організму на його ферментний апарат особливо наочно просліджується у мікроорганізмів. Наприклад, кишкова паличка при зростанні на живильному середовищі, що містить глюкозу, синтезує лише сліди b-галактозідази. У присутності ж різних b-галактозідів утворюються значні кількості цього Ф. – до 6–7% від всіх білків, що містяться в клітці. Ф., новоутворення або посилення синтезу яких відбувається під впливом якого-небудь з'єднання, називаються індукованими ферментами . Під впливом ін. з'єднань може відбуватися придушення синтезу Ф., зване репресією. У тваринному організмі індукція і репресія синтезу Ф. здійснюється не лише під впливом відповідних субстратів і метаболітов, але і під впливом гормонів. Так, синтез глюкозо-6-фосфатазы, що бере участь в синтезі глюкози в печінці, індукується гормонами тіроксином і кортизоном, але репресується інсуліном. Загальна теорія індукції і репресії біосинтезу на генетичному рівні дана французькими ученими Ф. Жакобом і Ж. Моно (див. Оперон ) . У одному організмі один і той же Ф. може бути представлений різними молекулярними формами. Такі всілякі форми Ф., що каталізують одну і ту ж реакцію, але що розрізняються по фізичних, хімічних і імунологічних властивостях, називаються ізоферментами . Синтез ізоферментів визначається генетичними чинниками, але може змінюватися під впливом умов існування організму. Т. о., чинники, від яких залежать концентрація і активність Ф. у організмі, так само всілякі, як і умови його існування. Це перш за все водний, газовий, температурний, кислотний і світловий режим середовища, а також концентрація субстратів і різних кофакторів, необхідних для дії Ф., наявність активаторів і інгібіторів, концентрації метаболітов і, нарешті, у вищих багатоклітинних організмів це нервова і гормональна регуляція ферментативної активності.
Прикладом впливу умов існування організму на активність Ф. може служити Пастера ефект – припинення бродіння під дією кисню. Активність багатьох Ф. регулюється за аллостерічеському принципом. В таких Ф. є т.з. аллостерічеський центр, приєднуючись до якого певний метаболіт – еффектор викликає зміну структури активного центру, унаслідок чого активність Ф. знижується або підвищується.
Деякі Ф. знаходяться в клітці у вигляді багатоферментних комплексів. У таких багатоферментних ансамблях активність кожного окремого Ф. строго координована і регулюється ін. Ф., що входять до складу даного комплексу. Прикладом багатоферментного комплексу може служити піруватдегидрогеназа, що складається з 16 молекул піруватдекарбоксилази, 8 молекул дігидроліпоїлдегидрогенази і 4 агрегатів ліпоатацетілтрансферази, кожна з яких складається з 16 субодиниць. Вирішальну роль в регуляції активності Ф. у клітці грають різні субклітинні структури – мітохондрії, мікросоми, лізосоми і т.д., і белковоліпідниє мембрани, що відокремлюють їх від цитоплазми. Багато Ф. вмонтовані в цих мембранах у вигляді багатоферментних ансамблів.
Практичне значення ферментів. Ферментативні процеси є основою багатьох виробництв: хлібопечення, виноробства, пивоваріння, сироваріння, виробництва спирту, сподіваючись, оцту. З початку 20 ст за пропозицією япон.(японський) ученого Д. Такаміне в спиртній і ін. галузях промисловості почалося вживання ферментних препаратів, що отримуються з плісневих грибів або бактерій. У ряді країн цей спосіб широко використовується для зцукрення за допомогою амілаз крохмалистої сировини з метою здобуття кристалічної глюкози або його зброджування на спирт. Концентровані амілолітичні препарати Ф. з плісневих грибів при добавці в тісто приводять до поліпшення якості хліба і прискорення технологічного процесу. Препарати протеолітичних Ф., отримуваних з мікроорганізмів, уживаються в шкіряній промисловості для видалення волосся і м'якшення сировини, а в сироварній промисловості – для заміни дефіцитного сичужного ферменту (ренніну ) . Препарати мікробних пектолітичних Ф. широко використовують при виробництві соків (вихід плодового соку підвищується на 10–20%). Все більше вживання очищені ферментні препарати знаходять в медицині. У наукових дослідженнях і в клінічній практиці високоочищені ферментні препарати служать як специфічні засоби біохімічного аналізу (див. Ферментативні методи аналізу ) . Вельми перспективне вживання т.з. іммобілізованних Ф., які зв'язуються яким-небудь носієм, створюючим з даним Ф. нерозчинний комплекс. При підборі відповідного носія можна отримати іммобілізованний Ф. з високою активністю, стійкий по відношенню до денатуруючих агентів. Колонка, заповнена іммобілізованним Ф., може бути багато разів використана для проведення відповідної реакції. Іммобілізованниє Ф. знаходять усе більш широке вживання в аналітичній практиці і біохімічній технології.
Літ.: Ферменти, М., 1964; Діксон М., Уебб Е., Ферменти, пер.(переведення) з англ.(англійський), М., 1966; Номенклатура ферментів, пер.(переведення) з англ.(англійський), М., 1966; Бернхард С., Структура і функція ферментів, пер.(переведення) з англ.(англійський), М., 1971; Структура і функція ферментів, ст 1–2, М., 1972–73; Фениксова Р. Ст, Біохімічні основи здобуття і вживання ферментних препаратів, в кн.: Технічна біохімія, М., 1973; Кретовіч Ст Л., Введення в ензімологию, 2 видавництва, М-коди,, 1974; Аллостерічеськие ферменти, М., 1975; Ферменти медичного призначення, Л., 1975; Ферментні препарати в харчовій промисловості, М., 1975; Advances in enzymology and related areas of molecular biology, v. 1–43, N. Y., 1941–75; Methods in enzymology, v. 1–36, N. Y., 1955–75.