Каталіз (від греч.(грецький) katálysis — руйнування), зміна швидкості хімічних реакцій у присутності речовин> (каталізаторів ), вступаючих в проміжну хімічну взаємодію з реагуючими речовинами, але поновлюючих після кожного циклу проміжних взаємодій свій хімічний склад. Реакції за участю каталізаторів називаються каталітичними. Кількість реагуючої речовини, яка може випробувати перетворення у присутності певної кількості каталізатора, не обмежується якими-небудь стехіометричними співвідношеннями і може бути дуже великим. Цим каталітичні реакції відрізняються від індукованих, або зв'язаних реакцій, коли одна реакція викликається або прискорюється (індукується) іншій і відбувається необоротне перетворення речовини-індуктора. Можливі зміни каталізатора при каталітичних реакціях є результатом побічних процесів, що аж ніяк не обумовлюють каталітичну дію.
Дію каталізатора відкриває нова реакційна дорога, зазвичай з великим числом стадій, на якому каталізатор входить до складу активного комплексу (активованого комплексу ) принаймні одній із стадій. Якщо при цьому швидкість реакції стає більше, ніж у відсутність каталізатора, то До. називається позитивним (його незрідка ототожнюють із загальним поняттям До.). Можливий і зворотний випадок, коли відбувається негативний К.: у присутності каталізатора виключається одна з можливих доріг реакції і залишаються лише повільніші, внаслідок чого реакція сповільнюється або навіть практично повністю пригнічується (див. Антиокислювачі, Інгібітори хімічні ). Особливий випадок До. — прискорення реакції при дії продукту реакції або однієї з проміжних речовин, що утворюються при реакції (див. Автокаталіз ).
До. не пов'язаний із зміною вільній енергії каталізатора, і дія каталізатора не може тому зміщувати положення рівноваги хімічної реакції. Поблизу стану рівноваги каталізатори в рівній мірі прискорюють як пряму, так і зворотну реакцію.
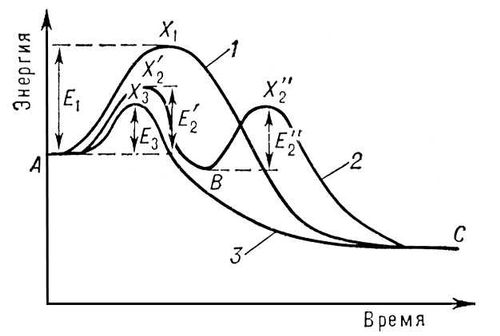
Основним чинником, що визначає швидкість хімічного перетворення, є енергія активації ( Е ) — різниця енергій активного комплексу і вихідних реагуючих молекул. Якщо передбачити, що реакція не порушує рівноважного розподілу енергії між молекулами, то вірогідність утворення активного комплексу, а отже, і швидкість реакції в першому наближенні пропорційна exp (— E/rt ), де R — газова постійна, Т — абсолютна температура. Звідси витікає, що швидкість реакції тим більше, чим менше Е , і унаслідок експоненціальної залежності зростає значно навіть при невеликому зниженні Е . На мал. представлена зміна енергії при реакції без каталізатора (крива 1 ) і за участю каталізатора (криві 2 і 3 ). Крива 2 з двома максимумами відповідає утворенню одного проміжного продукту. Число стадій і проміжних продуктів часто буває значно великим. Взаємодія реагуючих речовин з каталізатором може і не приводити до утворення стабільної форми проміжного з'єднання (крива 3 ). Але і в цьому випадку каталізатор входить до складу активного комплексу і взаємодія реагуючих речовин з каталізатором визначає реакційну дорогу. Якщо енергії активних комплексів всіх стадій реакційної дороги за участю каталізатора нижче за енергію активного комплексу реакції без каталізатора (тобто>, і E 3 нижче E 1 ), то участь каталізатора приведе до збільшення швидкості (позитивний До.). У багатьох випадках До. прискорення реакції досягається завдяки появі багатих енергією часток в процесі самої реакції, причому їх концентрація може перевершувати рівноважну (див. Ланцюгові реакції ). Наприклад, каталітична дія води на окислення окислу вуглецю пов'язана з розвитком реакційних доріг за участю гідроксильних груп і атомів водню. Негативний До. часто пов'язаний з припиненням ланцюгової реакції унаслідок обриву ланцюгів при взаємодії негативного каталізатора з активними частками. Прикладом може служити уповільнюючий вплив кисню на з'єднання водню з хлором.
Характер проміжної хімічної взаємодії при До. вельми всілякий. Зазвичай розрізняють дві групи каталітичних процесів: кислотно-основний (гетеролітичний) і окислювально-відновний (гомолітічеський). У процесах першої групи відбувається проміжна кислотно-основна взаємодія реагуючих речовин з каталізатором наприклад перехід протона від каталізатора до реагуючих речовин або навпаки. На подальших стадіях протон переміщається у зворотному напрямі, і каталізатор відновлює свій склад. При До. апротонними кислотами взаємодія здійснюється через вільну пару електронів реагуючої речовини. Прикладами кислотно-основного До. можуть служити гідроліз складних ефірів, що прискорюється кислотами; гідратація олефінов у присутності фосфорно-кислотних каталізаторів; ізомеризація і крекінг вуглеводнів на алюмосилікатних каталізаторах; алкилірованіє; полімеризація і багато інших реакцій. При реакціях окислювально-відновного До. проміжна взаємодія пов'язана з електронними переходами між каталізатором і реагуючими речовинами. До цієї групи відносяться окислення двоокису сірки в триокис у виробництві сірчаної кислоти; окислення аміаку до окислу азоту при здобутті азотної кислоти; багаточисельні процеси парціального окислення органічних сполук, наприклад етилену в окисел етилену, нафталіну у фталевий ангідрид; гідрогенізація; дегідрогенізація; циклізація і ароматизація вуглеводнів; розкладання перекису водню і багато ін. Каталітичною активністю відносно окислювально-відновних реакцій володіють переважно метали 4-, 5- і 6-го періодів системи Д. І. Менделєєва, що мають недобудовану d -оболочку електронів, їх з'єднання і в меншій мірі з'єднання елементів з що добудовується f -оболочкой (лантаноїди і актиноїди).
Розглянуті групи далеко не охоплюють всю різноманітність каталітичних реакцій. Характер проміжної взаємодії при До. набагато складніший і залежить від всіх деталей електронної структури як реагуючих речовин, так і каталізатора. Конкретні механізми каталітичних реакцій багатообразні і доки лише в небагатьох випадках твердо встановлені.
В залежності від фазового стану реагуючих речовин і каталізатора розрізняють гомогенний і гетерогенний К. Промежуточноє положення займає мікрогетерогенний До. у колоїдних системах (наприклад, До. ферментами). При гомогенному До. каталізатор і реагуючі речовини утворюють одну однорідну систему, кордони розділу між каталізатором і реагуючими речовинами відсутні. При гетерогенному До. каталізатор і реагуючі речовини знаходяться в різних фазах і відокремлені один від одного кордоном розділу. Найбільш важливі випадки, коли каталізатор є твердим тілом, а реакційна система утворює рідку або газоподібну фазу. Проміжна взаємодія відбувається при цьому переважно на поверхні твердого каталізатора.
Вибір складу каталізатора для певної реакції є дуже складною проблемою, що вирішується доки головним чином емпіричним дорогою. У СРСР запропонований і розвинений ряд теоретичних підходів, заснованих на кореляції окремих приватних властивостей каталізаторів з їх активністю. Так, мультіплетная теорія До. (перші публікації 1929) передбачає проміжну взаємодію реагуючих речовин з декількома атомами на поверхні твердих каталізаторів і надає вирішальне значення відповідності відстаней між атомами в молекулах реактантов і параметрів кристалічної структури каталізатора. Надалі теорія була доповнена уявленням про необхідність певної відповідності енергій зв'язків, що розриваються і утворюються в результаті реакції, і енергій зв'язків реактантов з каталізатором при проміжній взаємодії. Значне поширення в 50-х рр. отримало уявлення про залежність каталітичної активності твердих каталізаторів, що володіють напівпровідниковими властивостями, від їх електричних характеристик, — так звана електронна теорія К. По цій теорії передбачається, що проміжна взаємодія реактантов з каталізатором здійснюється за участю електронів провідності твердого каталізатора і тому залежить від його колективних електронних властивостей — розташування енергетичних зон і локальних рівнів електронів, роботи виходу електрона, концентрації носіїв струму і ін. У гетерогенному До. широко використовувалося припущення (висунуте в 1939) про існування на поверхні твердих каталізаторів особливих активних центрів, що є ребрами, кутами або різними структурними порушеннями (дислокації) нормальної кристалічної структури. Передбачалося також, що при нанесенні каталітично активної речовини на інертний носій особливі каталітичні властивості проявляють окремо розташовані атоми або сукупності невеликого числа атомів — ансамблі.
Поява точних методів визначення поверхні каталізаторів дозволила встановити, що активність, віднесена до одиниці поверхні (питома каталітична активність) визначається хімічним складом і дуже мало залежить від структурних дислокацій. Питома каталітична активність різних граней кристалів інколи розрізняється у декілька разів. Великий вплив на активність роблять порушення хімічного складу (відхилення від стехиометрії, впровадження домішок, локальні хімічні утворення і тому подібне).
В 60-і роки проміжна хімічна взаємодія в гетерогенному До. розглядається переважно як локальне, визначуване електронною структурою окремих атомів або іонів каталітично активного компонента на поверхні каталізатора з врахуванням впливу найближчого оточення. Значну допомогу в розвитку цього підходу надала виявлена експериментально аналогія у дії твердих каталізаторів, що містять певний метал, при гетерогенному До. і розчинних комплексів, компонентом яких є той же метал, при гомогенному До. у розчинах. При цьому використовуються теорії кристалічного поля і поля лігандов, що ще раніше успішно застосовувалися в хімії комплексних з'єднань. Для ряду класів каталізаторів і каталітичних реакцій встановлені кореляції між каталітичною активністю і енергіями зв'язків реактантов з каталізатором при проміжній взаємодії, що полегшують в окремих випадках підбір каталізаторів.
Перші наукові відомості про До. відносяться до початку 19 ст У 1806 французькі хіміки Н. Клеман і Ш. Дезорм відкрили каталітичну дію оксидів азоту на окислення сірчистого газу в камерному процесі здобуття сірчаної кислоти, В 1811 російський хімік К. С. Кирхгоф відкрило, що розбавлені кислоти здатні викликати перетворення крохмалю на цукор (глюкозу); у 1814 їм же було встановлено, що цю реакцію може каталізувати діастази з ячмінного солоду, — так належало початок вивченню біологічних каталізаторів — ферментів. У 1818 французький хімік Л. Тенар встановив, що велике число твердих тіл надає прискорююча дія на розкладання розчинів перекису водню, а англійський хімік Г. Деві відкрив здатність пари спирту і ефіру окислюватися киснем на платині. У 1822 йому.(німецький) хімік І. Деберейнер встановив, що водень і кисень з'єднуються на платині при звичайній температурі. За цим послідувало відкриття і ряду ін. прикладів різкої позитивної дії речовин на швидкість або виникнення хімічних реакцій. Це привело до виділенню особливої групи явищ, названих йому.(німецький) хіміком Е. Мічерліхом контактними (1833) і швед.(шведський) хіміком І. Берцеліусом каталітичними (1835).
Надалі було відкрите велике число каталітичних реакцій, і за останніх 50 років До. став провідним методом здійснення хімічних реакцій в промисловості. Вживання каталізаторів дозволяє проводити хімічні перетворення з високими швидкостями при невеликих температурах — більшість промислових каталітичних процесів без каталізаторів взагалі не могли б бути реалізоване. Підбираючи каталізатори, можна направляти хімічні перетворення у бік освіти певного продукту з ряду можливих. Вживання стереоспецифічних каталізаторів дозволяє регулювати і будову кінцевих продуктів, наприклад полімерів. З допомогою До. на початку 20 ст була вирішена проблема фіксації азоту повітря. Промоутовані залізні і інші каталізатори дозволили здолати хімічну інертність елементарного азоту і здійснити синтез аміаку. Одночасно був розроблений каталітичний метод здобуття азотної кислоти шляхом окислення аміаку на платинових сітках. На каталітичних реакціях грунтуються сучасні методи здобуття водню з природного газу. Каталітичні методи займають пануюче положення і в технології нафтопереробки. Сотні мільйонів тонн високоякісного моторного палива виробляються з допомогою каталітичних реакцій крекінгу, гідрокрекінгу, ріформінга, циклізації і ізомеризації вуглеводнів нафти. Особливо велику роль грають каталітичні методи в здійсненні процесів органічного синтезу. У нашій країні вперше в світі було розроблене і реалізоване виробництво синтетичного каучуку, засноване на перетворенні етилового спирту на дивініл за допомогою багатокомпонентного окисного каталізатора Лебедева. Каталітичні методи використовуються для здобуття переважної більшості продуктів нафтохімічного синтезу: розчинників, ароматичних вуглеводнів, мономерів для виробництва синтетичних каучуків, синтетичних волокон і ін. полімерних матеріалів. Каталізатори широко використовуються і для полімеризації.
До. грає провідну роль в хімічних перетвореннях в живій природі. Вся складна система управління життєвими процесами в організмах заснована на каталітичних реакціях. Біологічні каталізатори, звані ферментами або ензимами, є речовинами білкової природи з хімічно активними групами, що часто включають в свій склад атоми перехідних елементів. По деяких властивостях ферменти перевершують промислові каталізатори. У СРСР і за кордоном широко ведуться дослідження нових типів складних синтетичних каталізаторів — комплексних з'єднань, органічних напівпровідників, полімерів, що характеризуються простішим складом в порівнянні з ферментами, але що моделюють до певної міри їх дію. Науці про До. належить істотна роль як в прогресі хімічної промисловості, так і в розкритті найважливіших біологічних закономірностей.
Літ.: Баландін А. А., Мультіплетная теорія каталізу, ч, 1—2, М., 1963—64; Волькенштейн Ф. Ф., Електронна теорія каталізу на напівпровідниках, М., 1960: Catalysis, ed. P. Н. Ernmett, v. 1—7, N. Y., 1954—60; Ашмор П.., Каталіз і інгібірування хімічних реакцій, пер.(переведення) з англ.(англійський), М., 1966; Томас Дж., Томас В., Гетерогенний каталіз, пер.(переведення) з англ.(англійський). М., 1969; Киперман С. Л., Введення в кінетику гетерогенних каталітичних реакцій, М., 1964; Бореськов Р. До., Каталіз у виробництві сірчаної кислоти, М. — Л., 1954; Крилов О. Ст, Каталіз неметалами, Л., 1967; Основи передбачення каталітичної дії. Праці IV Міжнародного конгресу з каталізу, т. 1—2, М., 1970.