Спадкові захворювання, хвороби, обумовлені порушеннями в процесах зберігання, передачі і реалізації генетичної інформації. З розвитком генетики людини, у тому числі і генетики медичної, з'ясувалася спадкова природа багатьох захворювань і синдромів, що вважалися раніше хворобами з невстановленою етіологією. Роль спадкових чинників підтверджується більш високою частотою ряду захворювань в деяких сім'ях в порівнянні з населенням в цілому. Вивченням Н. з. людини займається переважно медична генетика.
В основі Н. з. лежать мутації — переважно хромосомні і генні, відповідно чому умовно говорять про хромосомних хворобах і власне спадкових (генних) хворобах. Мутація веде до порушення синтезу певного поліпептиду (структурного білка або ферменту). Залежно від того, яка роль цього поліпептиду в життєдіяльності організму, у хворого виникають порушення (зміни фенотипа ) локального або системного порядку. Найбільш раціональна класифікація Н. з. по характеру метаболічних розладів: порушення обміну амінокислот (приклади: фенілпіровиноградна олігофренія, тірозіноз, алкаптонурія); порушення обміну ліпідів (хвороба Німана — Спис, хвороба Гоші); порушення обміну вуглеводів (галактоземія, фруктозурія); порушення мінерального обміну (гепатоцеребральная дистрофія ); порушення білірубінового обміну (синдром Кріглер — Нацжара, синдром Дубініна — Джонсона). (Див. також «Молекулярні хвороби» .) Проте оскільки біохімічні механізми більшості Н. з. поки невідомі, і, отже, патогенетична класифікація ще не може бути повною, її доповнюють класифікацією за органно-системним принципом: Н. з. крові (гемолітична хвороба новонароджених, гемоглобінопатії ); ендокринної системи (адреногенітальний синдром, діабет цукровий ); Н. з. з переважним ураженням нирок (фосфат-діабет, цистиноз); сполучній тканині (хвороба Марфана, мукополісахарідози); нервово-м'язової системи (прогресуюча м'язова дистрофія) і т.д.
Залежно від того, де локалізований патологічний (мутант) ген — в аутосомі або в статевій хромосомі — і які його взаємини з нормальним аллелем, тобто чи є мутація домінантною (нормальний ген пригнічується патологічним) або рецесивною (патологічний ген пригнічується нормальним), розрізняють наступні основні типи спадкоємства: аутосомно-домінантний, аутосомно-рецесивний і зчеплений з підлогою (або обмежений підлогою). Тип спадкоємства встановлюється шляхом аналізу родоводу. При складанні останньою враховуються поширення в сім'ї захворювання, що вивчається, і родинного відношення між хворими. Побудова і аналіз родоводу складають предмет клінико-генеалогічного дослідження.
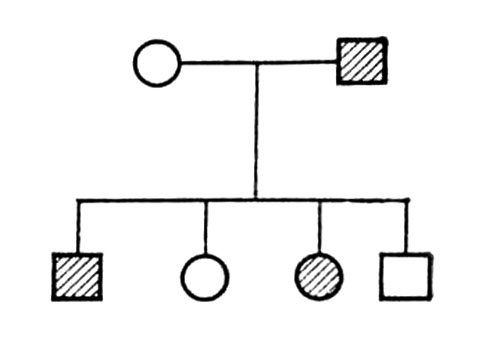
При захворюваннях, успадкованих за аутосомно-домінантним типом ген мутанта виявляється вже в гетерозиготному стані (див. Гетерозиготність ); хворі хлопчики і дівчатка народжуються з однаковою частотою; патологічна спадковість просліджується в родоводі «по вертикалі»; принаймні один з батьків хворого також хворий. Родовід, характерний для аутосомно-домінантного типа спадкоємства, представлений на мал.(малюнок) 1.
За аутосомно-домінантним типом успадковуються, наприклад, арахнодактілія, ахондроплазія, брахідактилія, геморагічна телеангіоектазія Ослера, гіпербілірубінемія, нейрофіброматоз Реклінгаузена, пельгеровськая аномалія лейкоцитів, полідактилія, птоз спадковий, пурпуру тромбоцитопенічна ідіопатична, ектопія кришталика і ін.
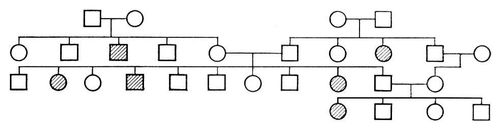
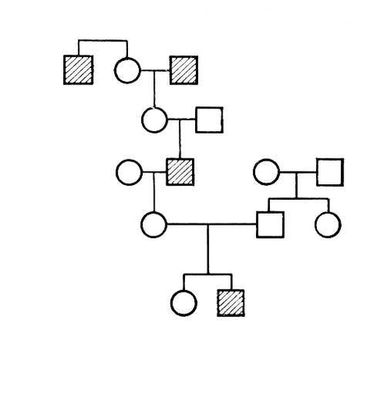
При захворюваннях, успадкованих за аутосомно-рецесивним типом, ген мутанта виявляється лише в гомозиготному стані (див. Гомозиготність ); хворі хлопчики і дівчатка народжуються з однаковою частотою; батьки хворих фенотіпічеськи здорові, але є гетерозиготними носіями гена мутанта; патологічна спадковість просліджується в родоводі сім'ї «по горизонталі»; вірогідність народження хворих дітей зростає в разі кровної спорідненості батьків. Родовід, характерний для аутосомно-рецесивного типа спадкоємства, представлений на мал.(малюнок) 2. Якщо один з батьків гомозіготен по патологічному рецесивному гену, а інший є його гетерозиготним носієм, то в половині випадків діти можуть виявитися хворими, і створюється враження спадкоємства захворювання за домінантним типом (родовід на мал.(малюнок) 3 ). Таке явище носить назва псевдодомінування. Від дійсного домінування воно відрізняється тим, що хворі з рецесивною мутацією в браку із здоровими людьми завжди даватимуть здорове потомство, а здорові в браку з гетерозиготними носіями з певною частотою (25%) матимуть хворих дітей. Якщо прослідити родовід, представлений на мал. 3 , ще в одному поколінні, вона може виглядати, наприклад, як на мал. 4 . За аутосомно-рецесивним типом успадковуються агамаглобулінемія, алкаптонурія, альбінізм, амавротічеськая ідіотія, гепатоцеребральная дистрофія, дистонія м'язова деформує, муковісцидоз, серповидноклітинна анемія і ін.
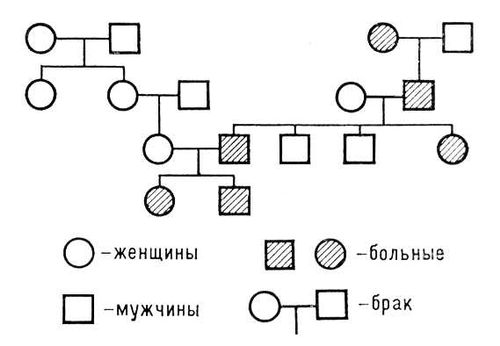
Із захворювань зчеплених з підлогою або обмежених підлогою, для клініки особливе значення мають хвороби, обумовлені рецесивними мутаціями в Х-хромосомі (цей тип спадкоємства називається також Х-хромосомнім). Жінки з такого типа мутацією, як правило, фенотіпічеськи здорові, оскільки рецесивному патологічному гену протистоїть у них нормальний аллель іншої Х-хромосоми. У чоловіків же ген мутанта представлений в однині і визначає патологію фенотипа. При хворобах що передаються за Х-хромосомнім типом, дія гена мутанта виявляється лише в гетерогаметного підлоги (тобто у чоловіків); у обтяжених сім'ях захворює половина синів, а половина дочок — носії гена мутанта (кондуктори); батьки клінічно здорові. Хвороба часто виявляється у синів сестер хворого (пробанду) або у його двоюрідних братів по материнській лінії. Хворий батько не передає дефектний ген синам. Типовий родовід представлений на мал.(малюнок) 5.
За Х-хромосомнім типом успадковуються гемофілія А, гемофілія В, періодичний параліч, пігментний ретиніт, фосфат-діабет, колірна сліпота і ін.
Перераховані типи спадкоємства передбачають головним чином моногенні захворювання (визначувані мутацією одного гена). Проте патологічний стан може залежати від двох і генів більш мутантів. Ряд патологічних генів володіє пониженою пенетрантністю . При цьому присутність їх в геномі, навіть в гомозиготному стані, необхідно, але недостатньо для розвитку хвороби. Т. о., не всі типи спадкоємства хвороб людини укладаються в перераховані схеми.
Оскільки всякий фенотип, як нормальний, так і патологічний, детермінується не лише генотипом і є результатом взаємодії генотипу і середовища, остільки спадковій патології властивий значний клінічний поліморфізм: в межах однієї нозологічної одиниці можуть зустрічатися різні клінічні синдроми, міра тягаря захворювання також варіює в широких межах. Велика варіабельность клінічних проявів і перебігу Н. з. спостерігається деколи навіть у членів однієї сім'ї. Для об'єктивної оцінки співвідносної ролі спадкових чинників і середовища в етіології і патогенезі Н. з. поважно вивчати особливості їх клінічної картини і течії в однояйцевих і різнояєчних близнят .
Нозологічна приналежність Н. з. встановлюється на основі всестороннього клінічного (у тому числі клінико-генеалогічного) і лабораторного обстеження. Велику діагностичну цінність мають біохімічні, електрофізіологічні, цитоморфологичеськие імунологічні і ін. лабораторні методи, що часто дозволяють ідентифікувати не лише захворювання, але і гетерозиготне носійство гена мутанта. Інколи діагностику полегшує плейотропний ефект генів, т. е, множинність залежних від них фенотипічних проявів. Зокрема, дія патологічного гена може виявитися не лише в захворюванні, але і в ряду інших, зазвичай індиферентних для організму ознак, по яких в сумнівних випадках і встановлюється присутність гена-«віновника».
Завдяки прогресу медичної генетики і розширенню уявлень про характер спадкоємства різних захворювань і вплив чинників зовнішнього середовища на проявляємость генів мутантів стали набагато яснішими за дорогу лікування і профілактики Н. з. Основні принципи лікування: виключення або обмеження продуктів, перетворення яких в організмі у відсутності необхідного ферменту приводять до патологічного стану; заместітітельная терапія дефіцитним ферментом або нормальним кінцевим продуктом збоченої реакції; індукція дефіцитних ферментів. Велике значення надається чиннику своєчасності терапії, яку слід починати до розвитку в хворих виражених порушень. Деякі біохімічні дефекти можуть з віком частково компенсуватися. У перспективі великі надії покладаються на генну інженерію, під якою мається на увазі направлене втручання в структуру і функціонування генетичного апарату — видалення або виправлення генів мутантів, заміна їх нормальними.
Найважливішим завданням медичної генетики залишається профілактика Н. з., здійснювана в основному через медіко-генетічні консультації .
Літ.: Давіденков С. Н., Спадкові хвороби нервової системи, 2 видавництва, М., 1932; його же, Генетика медична, в кн.: Велика медична енциклопедія, 2 видавництва, т. 6, М., 1958; Довідник по клінічній генетиці, під ред. Л. О. Бадаляна, М., 1971; Schreier До., Die angeborenen Stoffwechselanomalien, Stuttg., 1963; Mc Kusick V. A., Mendelian inheritance in man, Bait., [1966]; The metabolic basis of inherited disease, ed. J. B. Stanbury [а. о.], N. Y. — [а. о.], 1972.