Наследственные заболевания, болезни, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации. С развитием генетики человека, в том числе и генетики медицинской, выяснилась наследственная природа многих заболеваний и синдромов, считавшихся ранее болезнями с неустановленной этиологией. Роль наследственных факторов подтверждается более высокой частотой ряда заболеваний в некоторых семьях по сравнению с населением в целом. Изучением Н. з. человека занимается преимущественно медицинская генетика.
В основе Н. з. лежат мутации — преимущественно хромосомные и генные, соответственно чему условно говорят о хромосомных болезнях и собственно наследственных (генных) болезнях. Мутация ведёт к нарушению синтеза определенного полипептида (структурного белка или фермента). В зависимости от того, какова роль этого полипептида в жизнедеятельности организма, у больного возникают нарушения (изменения фенотипа) локального или системного порядка. Наиболее рациональна классификация Н. з. по характеру метаболических расстройств: нарушения обмена аминокислот (примеры: фенилпировиноградная олигофрения, тирозиноз, алкаптонурия); нарушения обмена липидов (болезнь Нимана — Пика, болезнь Гоше); нарушения обмена углеводов (галактоземия, фруктозурия); нарушения минерального обмена (гепатоцеребральная дистрофия); нарушения билирубинового обмена (синдром Криглер — Нацжара, синдром Дубинина — Джонсона). (См. также «Молекулярные болезни».) Однако поскольку биохимические механизмы большинства Н. з. пока неизвестны, и, следовательно, патогенетическая классификация ещё не может быть полной, её дополняют классификацией по органно-системному принципу: Н. з. крови (гемолитическая болезнь новорождённых, гемоглобинопатии); эндокринной системы (адреногенитальный синдром, диабет сахарный); Н. з. с преимущественным поражением почек (фосфат-диабет, цистиноз); соединительной ткани (болезнь Марфана, мукополисахаридозы); нервно-мышечной системы (прогрессирующие мышечные дистрофии) и т.д.
В зависимости от того, где локализован патологический (мутантный) ген — в аутосоме или в половой хромосоме — и каковы его взаимоотношения с нормальным аллелем, т. е. является ли мутация доминантной (нормальный ген подавляется патологическим) или рецессивной (патологический ген подавляется нормальным), различают следующие основные типы наследования: аутосомно-доминантный, аутосомно-рецессивный и сцепленный с полом (или ограниченный полом). Тип наследования устанавливается путём анализа родословной. При составлении последней учитываются распространение в семье изучаемого заболевания и родственного отношения между больными. Построение и анализ родословной составляют предмет клинико-генеалогического исследования.
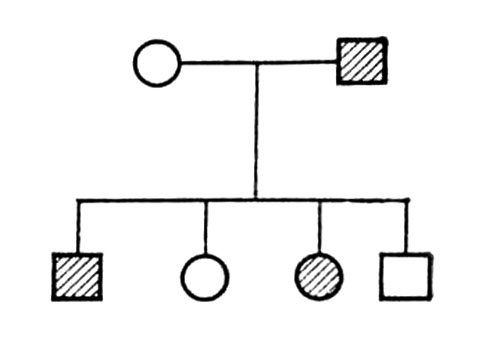
При заболеваниях, наследуемых по аутосомно-доминантному типу, мутантный ген проявляется уже в гетерозиготном состоянии (см. Гетерозиготность); больные мальчики и девочки рождаются с одинаковой частотой; патологическая наследственность прослеживается в родословной «по вертикали»; по крайней мере один из родителей больного также болен. Родословная, характерная для аутосомно-доминантного типа наследования, представлена на рис.(рисунок) 1.
По аутосомно-доминантному типу наследуются, например, арахнодактилия, ахондроплазия, брахидактилия, геморрагическая телеангиэктазия Ослера, гипербилирубинемия, нейрофиброматоз Реклингаузена, пельгеровская аномалия лейкоцитов, полидактилия, птоз наследственный, пурпура тромбоцитопеническая идиопатическая, эктопия хрусталика и др.
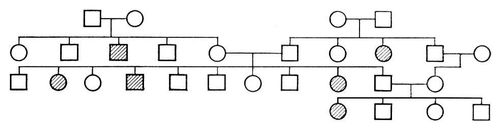
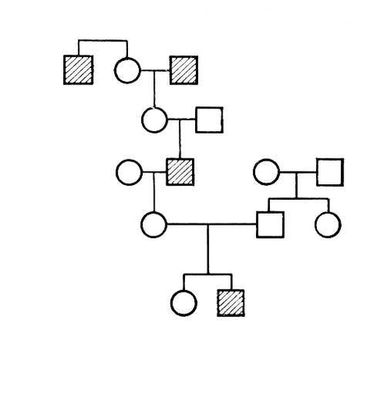
При заболеваниях, наследуемых по аутосомно-рецессивному типу, мутантный ген проявляется лишь в гомозиготном состоянии (см. Гомозиготность); больные мальчики и девочки рождаются с одинаковой частотой; родители больных фенотипически здоровы, но являются гетерозиготными носителями мутантного гена; патологическая наследственность прослеживается в родословной семьи «по горизонтали»; вероятность рождения больных детей возрастает в случае кровного родства родителей. Родословная, характерная для аутосомно-рецессивного типа наследования, представлена на рис.(рисунок) 2. Если один из родителей гомозиготен по патологическому рецессивному гену, а другой является его гетерозиготным носителем, то в половине случаев дети могут оказаться больными, и создаётся впечатление наследования заболевания по доминантному типу (родословная на рис.(рисунок) 3). Такое явление носит название псевдодоминирования. От истинного доминирования оно отличается тем, что больные с рецессивной мутацией в браке со здоровыми людьми всегда будут давать здоровое потомство, а здоровые в браке с гетерозиготными носителями с определенной частотой (25%) будут иметь больных детей. Если проследить родословную, представленную на рис. 3, ещё в одном поколении, она может выглядеть, например, как на рис. 4. По аутосомно-рецессивному типу наследуются агаммаглобулинемия, алкаптонурия, альбинизм, амавротическая идиотия, гепатоцеребральная дистрофия, дистония мышечная деформирующая, муковисцидоз, серповидноклеточная анемия и др.
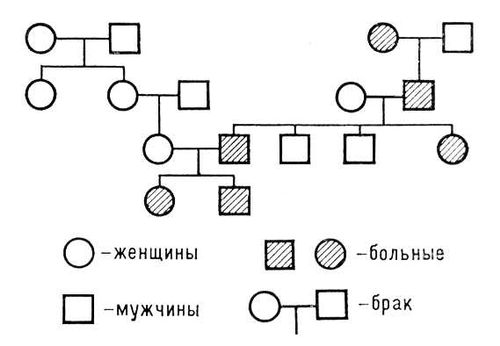
Из заболеваний, сцепленных с полом или ограниченных полом, для клиники особое значение имеют болезни, обусловленные рецессивными мутациями в Х-хромосоме (этот тип наследования называется также Х-хромосомным). Женщины с такого типа мутацией, как правило, фенотипически здоровы, поскольку рецессивному патологическому гену противостоит у них нормальный аллель другой Х-хромосомы. У мужчин же мутантный ген представлен в единственном числе и определяет патологию фенотипа. При болезнях, передающихся по Х-хромосомному типу, действие мутантного гена проявляется только у гетерогаметного пола (т. е. у мужчин); в отягощенных семьях заболевает половина сыновей, а половина дочерей — носители мутантного гена (кондукторы); родители клинически здоровы. Болезнь часто обнаруживается у сыновей сестёр больного (пробанда) или у его двоюродных братьев по материнской линии. Больной отец не передаёт дефектный ген сыновьям. Типичная родословная представлена на рис.(рисунок) 5.
По Х-хромосомному типу наследуются гемофилия А, гемофилия В, периодический паралич, пигментный ретинит, фосфат-диабет, цветовая слепота и др.
Перечисленные типы наследования предусматривают главным образом моногенные заболевания (определяемые мутацией одного гена). Однако патологическое состояние может зависеть от двух и более мутантных генов. Ряд патологических генов обладает сниженной пенетрантностью. При этом присутствие их в геноме, даже в гомозиготном состоянии, необходимо, но недостаточно для развития болезни. Т. о., не все типы наследования болезней человека укладываются в перечисленные схемы.
Поскольку всякий фенотип, как нормальный, так и патологический, детерминируется не только генотипом и является результатом взаимодействия генотипа и среды, постольку наследственной патологии присущ значительный клинический полиморфизм: в пределах одной нозологической единицы могут встречаться различные клинические синдромы, степень тяжести заболевания также варьирует в широких пределах. Большая вариабельность клинических проявлений и течения Н. з. наблюдается порой даже у членов одной семьи. Для объективной оценки соотносительной роли наследственных факторов и среды в этиологии и патогенезе Н. з. важно изучать особенности их клинической картины и течения у однояйцевых и разнояйцевых близнецов.
Нозологическая принадлежность Н. з. устанавливается на основе всестороннего клинического (в том числе клинико-генеалогического) и лабораторного обследования. Большую диагностическую ценность имеют биохимические, электрофизиологические, цитоморфологические, иммунологические и др. лабораторные методы, часто позволяющие идентифицировать не только заболевание, но и гетерозиготное носительство мутантного гена. Иногда диагностику облегчает плейотропный эффект генов, т. е, множественность зависящих от них фенотипических проявлений. В частности, действие патологического гена может проявиться не только в заболевании, но и в ряде других, обычно индифферентных для организма признаков, по которым в сомнительных случаях и устанавливается присутствие гена-«виновника».
Благодаря прогрессу медицинской генетики и расширению представлений о характере наследования различных заболеваний и влиянии факторов внешней среды на проявляемость мутантных генов стали намного яснее пути лечения и профилактики Н. з. Основные принципы лечения: исключение или ограничение продуктов, превращения которых в организме в отсутствии необходимого фермента приводят к патологическому состоянию; заместитительная терапия дефицитным ферментом или нормальным конечным продуктом извращённой реакции; индукция дефицитных ферментов. Большое значение придаётся фактору своевременности терапии, которую следует начинать до развития у больных выраженных нарушений. Некоторые биохимические дефекты могут с возрастом частично компенсироваться. В перспективе большие надежды возлагаются на генную инженерию, под которой подразумевается направленное вмешательство в структуру и функционирование генетического аппарата — удаление или исправление мутантных генов, замена их нормальными.
Важнейшей задачей медицинской генетики остаётся профилактика Н. з., осуществляемая в основном через медико-генетические консультации.
Лит.: Давиденков С. Н., Наследственные болезни нервной системы, 2 изд., М., 1932; его же, Генетика медицинская, в кн.: Большая медицинская энциклопедия, 2 изд., т. 6, М., 1958; Справочник по клинической генетике, под ред. Л. О. Бадаляна, М., 1971; Schreier К., Die angeborenen Stoffwechselanomalien, Stuttg., 1963; Mc Kusick V. A., Mendelian inheritance in man, Bait., [1966]; The metabolic basis of inherited disease, ed. J. B. Stanbury [a. o.], N. Y. — [a. o.], 1972.