Рентгенівський структурний аналіз, методи дослідження структури речовини по розподілу в просторі і інтенсивностям розсіяного на аналізованому об'єкті рентгенівського випромінювання. Р. с. а. поряд з нейтронографієй і електронографією є дифракційним структурним методом; у його основі лежить взаємодія рентгенівського випромінювання з електронами речовини, в результаті якої виникає дифракція рентгенівських променів . Дифракційна картина залежить від довжини хвилі використовуваних рентгенівських променів і будови об'єкту. Для дослідження атомної структури застосовують випромінювання з довжиною хвилі ~1, тобто порядку розмірів атомів. Методами Р. с. а. вивчають метали, сплави, мінерали, неорганічні і органічні з'єднання, полімери, аморфні матеріали, рідини і гази, молекули білків, нуклеїнових кислот і т.д. Найуспішніше Р. с. а. застосовують для встановлення атомної структури кристалічних тіл. Це обумовлено тим, що кристали володіють строгою періодичністю будови і є створеними самій природою дифракційні грати для рентгенівських променів.
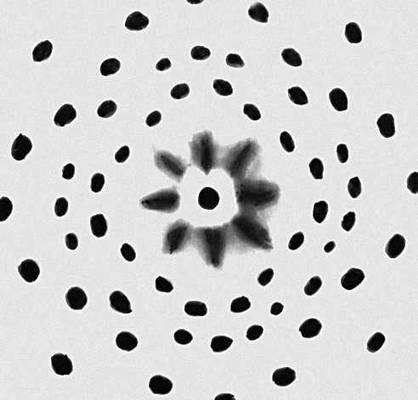
Історична довідка. Дифракція рентгенівських променів на кристалах була відкрита в 1912 німецькими фізиками М. Лауе, В. Фрідріхом і П. Кніппінгом. Направивши вузький пучок рентгенівських променів на нерухомий кристал, вони зареєстрували на поміщеній за кристалом фотопластині дифракційну картину, яка складалася з великого числа закономірно розташованих плям. Кожна пляма — слід дифракційного променя, розсіяного кристалом. Рентгенограма, отримана таким методом, носить назву лауеграмми ( мал. 1 ).
Розроблена Лауе теорія дифракції рентгенівських променів на кристалах дозволила зв'язати довжину хвилі l випромінювання, параметри елементарного вічка кристала а, b, з (див. Кристалічна решітка ) , кути падаючого (a 0 , b 0 , g 0 ) і дифракційного (а, b, g) променів співвідношеннями:
а (cosa— cosa 0 ) = h l,
b (cosb — cosb 0 ) = до l, (1)
з (cosg — cosg 0 ) = l l,
де h, до, I — цілі числа (міллеровськие індекси ) . Для виникнення дифракційного променя необхідне виконання приведених умов Лауе [рівнянь (1)], які вимагають, щоб в паралельних променях різниця ходу між променями, розсіяними атомами, що відповідають сусіднім вузлам грат, дорівнювали цілому числу довжин хвиль.
В 1913 В. Л. Брег і одночасно з ним Р. Ст Вульф запропонували наочніше трактування виникнення дифракційних променів в кристалі. Вони показали, що будь-яким з дифракційних променів можна розглядати як віддзеркалення падаючого променя від однієї з систем кристалографічної плоскості (дифракційне віддзеркалення, див.(дивися) Брега — Вульфа умова ) . В тому ж році В. Р. і У. Л. Брегги вперше досліджували атомні структури простих кристалів за допомогою рентгенівських дифракційних методів. У 1916 П. Дебай і німецький фізик П. Шеррер запропонували використовувати дифракцію рентгенівських променів для дослідження структури полікристалічних матеріалів. У 1938 французький кристалограф А. Гинье розробив метод рентгенівського малокутового розсіяння для дослідження форми і розмірів неоднородностей в речовині.
Застосовність Р. с. а. до дослідження широкого класу речовин, виробнича необхідність цих досліджень стимулювали розвиток методів розшифровки структур. У 1934 американський фізик А. Патерсон запропонував досліджувати будову речовин за допомогою функції міжатомних векторів (функції Патерсона). Американські учені Д. Харкер, Дж. Каспер (1948), В. Захаріасен, Д. Сейр і англійське вчене Ст Кокрен (1952) заклали основи так званих прямих методів визначення кристалічних структур. Великий внесок у розвиток патерсоновських і прямих методів Р. с. а. внесли Н. Ст Белов, Р. С. Жданов, А. І. Китайгородський, Би. До. Вайнштейн, М. Порай-Кошиц (СРСР), Л. Полінг, П. Евальд, М. Бюргер, Дж. Карле, Г. Хауптман (США), М. Вульфсон (Великобританія) і ін. Роботи по дослідженню просторової структури білка, початі в Англії Дж. Берналом (30-і рр.) і успішно продовжені Дж. Кендрю, М. Перуцем, Д. Кроуфут-Ходжкин і ін., зіграли виключно важливу роль в становленні молекулярній біології . В 1953 Дж. Уотсон і Ф. Крик запропонували модель молекули дезоксирибонуклеїнової кислоти (ДНК), яка добре узгоджувалася з результатами рентгенографічних досліджень ДНК(дезоксирибонуклеїнова кислота), отриманими М. Уїлкинсом .
В 50-х рр. почали бурхливо розвиватися методи Р. с. а. з використанням ЕОМ(електронна обчислювальна машина) в техніці експерименту і при обробці рентгенівської дифракційної інформації.
Експериментальні методи Р. с. а. Для створення умов дифракції і реєстрації випромінювання служать рентгенівські камери і рентгенівські дифрактометри . Розсіяне рентгенівське випромінювання в них фіксується на фотоплівці або вимірюється детекторами ядерних випромінювань . Залежно від стану досліджуваного зразка і його властивостей, а також від характеру і об'єму інформації, яку необхідно отримати, застосовують різні методи Р. с. а. Монокристали, що відбираються для дослідження атомної структури, повинні мати розміри ~ 0,1 мм і по можливості володіти досконалою структурою. Дослідженням дефектів в порівняно крупних майже досконалих кристалах займається рентгенівська топографія, яку інколи відносять до Р. с. а.
Метод Лауе — простий метод здобуття рентгенограм від монокристалів. Кристал в експерименті Лауе нерухомий, а використовуване рентгенівське випромінювання має безперервний спектр. Розташування дифракційних плям на лауеграммах ( мал. 1 ) залежить від симетрії кристала і його орієнтації відносно падаючого променя. Метод Лауе дозволяє встановити приналежність досліджуваного кристала до однієї і 11 лауевських груп симетрії і орієнтувати його (тобто визначати напрям кристалографічних осей) з точністю до декількох кутових хвилин. По характеру плям на лауеграммах і особливо появі астеризму можна виявити внутрішню напругу і деякі ін. дефекти кристалічної структури. Методом Лауе перевіряють якість монокристалів при виборі зразка для його повнішого структурного дослідження.
Методи гойдання і обертання зразка використовують для визначення періодів повторюваності (постійних грат) уздовж кристалографічного напряму в монокристалі. Вони дозволяють, зокрема, встановити параметри а , b, з елементарного вічка кристала. У цьому методі використовують монохроматичне рентгенівське випромінювання, зразок приводиться в коливальний або обертальний рух довкола осі, співпадаючої з кристалографічним напрямом, уздовж якого і досліджують період повторюваності. Плями на рентгенограмах гойдання і обертання, отриманих в циліндрових касетах, розташовуються на сімействі паралельних ліній. Відстані між цими лініями, довжина хвилі випромінювання і діаметр касети рентгенівської камери дозволяють обчислити шуканий період повторюваності в кристалі. Умови Лауе для дифракційних променів в цьому методі виконуються за рахунок зміни кутів, що входять в співвідношення (1) при гойданні або обертанні зразка.
Рентгенгоніометрічеськие методи. Для повного дослідження структури монокристала методами Р. с. а. необхідно не лише встановити положення, але і виміряти інтенсивності як можна більшого числа дифракційних віддзеркалень, які можуть бути отримані від кристала при даній довжині хвилі випромінювання і всіх можливих орієнтаціях зразка. Для цього дифракційну картину реєструють на фотоплівці в рентгенівському гоніометрі і вимірюють за допомогою мікрофотометра міра почорніння кожної плями на рентгенограмі. У рентгенівському дифрактометрі можна безпосередньо вимірювати інтенсивність дифракційних віддзеркалень за допомогою пропорційних, сцинтиляційних і інших лічильників рентгенівських квантів. Щоб мати повний набір віддзеркалень, в рентгенівських гоніометрах отримують серію рентгенограм. На кожній з них фіксуються дифракційні віддзеркалення, на міллеровськие індекси яких накладають певні обмеження (наприклад, на різних рентгенограмах реєструються віддзеркалення типа hk 0, hk 1 і так далі). Найчастіше виробляють рентгеногоніометрічеський експеримент по методах Вайсенберга. Бюргера ( мал. 2 ) і де Іонга — Боумена. Таку ж інформацію можна отримати і за допомогою рентгенограм гойдання.
Для встановлення атомної структури середньої складності (~ 50—100 атомів в елементарному вічку) необхідно виміряти інтенсивності декількох сотень і навіть тисяч дифракційних віддзеркалень. Ету вельми трудомістку і копітку роботу виконують автоматичні мікроденситометри і дифрактометри, керовані ЕОМ(електронна обчислювальна машина), інколи протягом декількох тижнів і навіть місяців (наприклад, при аналізі структур білків, коли число віддзеркалень зростає до сотень тисяч). Вживанням в дифрактометрі декількох лічильників, які можуть паралельно реєструвати віддзеркалення час експерименту удається значно скоротити. Дифрактометричні виміри перевершують фотореєстрацію по чутливості і точності.
Метод дослідження полікрісталлов (Дебая — Шеррера метод ) . Метали, сплави, кристалічні порошки складаються з безлічі дрібних монокристалів даної речовини. Для їх дослідження використовують монохроматичне випромінювання. Рентгенограма (дебаєграмма) полікрісталлов є декількома концентричними кільцями, в кожне з яких зливаються віддзеркалення від певної системи плоскості різно орієнтованих монокристалів. Дебаєграмми різних речовин мають індивідуальний характер і широко використовуються для ідентифікації з'єднань (у тому числі і в сумішах). Р.с.а. полікрісталлов дозволяє визначати фазовий склад зразків, встановлювати розміри і переважну орієнтацію (текстурування) зерен в речовині, здійснювати контроль за напругою в зразку і вирішувати інші технічні завдання.
Дослідження аморфних матеріалів і частково впорядкованих об'єктів. Чітку рентгенограму з гострими дифракційними максимумами можна отримати лише при повній тривимірній періодичності зразка. Чим нижче міра впорядкованості атомної будови матеріалу, тим більше розмитий, дифузний характер має розсіяне ним рентгенівське випромінювання. Діаметр дифузного кільця на рентгенограмі аморфної речовини може служити для грубої оцінки середніх міжатомних відстаней в нім. Із зростанням міри впорядкованості (див. Далекий порядок і ближній порядок ) в будові об'єктів дифракційна картина ускладнюється і, отже, містить більше структурної інформації.
Метод малокутового розсіяння дозволяє вивчати просторові неоднорідності речовини, розміри яких перевищують міжатомні відстані, тобто складають від 5—10 до ~ 10 000 . Розсіяне рентгенівське випромінювання в цьому випадку концентрується поблизу первинного пучка — в області малих кутів розсіяння. Малокутове розсіяння застосовують для дослідження пористих і мелкодісперсних матеріалів, сплавів і складних біологічних об'єктів: вірусів, клітинних мембран, хромосом. Для ізольованих молекул білка і нуклеїнових кислот метод дозволяє визначити їх форму, розміри, молекулярну масу; у вірусах — характер взаємного укладання складових їх компонент: білка, нуклеїнових кислот, ліпідів; у синтетичних полімерах — упаковку полімерних ланцюгів; у порошках і сорбентах — розподіл часток і пір по розмірах; у сплавах — виникнення і розміри фаз; у текстурах (зокрема, в рідких кристалах) — форму упаковки часток (молекул) в різного роду надмолекулярні структури. Рентгенівський малокутовий метод застосовується і в промисловості при контролі процесів виготовлення каталізаторів, високодисперсного вугілля і так далі Залежно від будови об'єкту виміру виробляють для кутів розсіяння від доль хвилини до декількох градусів.
Визначення атомної структури за даними дифракції рентгенівських променів. Розшифровка атомної структури кристала включає: встановлення розмірів і форми його елементарного вічка; визначення приналежності кристала до однієї з 230 федоровських (відкритих Е. С. Федоровим ) груп симетрії кристалів ; здобуття координат базисних атомів структури. Першу і частково другу завдання можна вирішити методами Лауе і гойдання або обертання кристала. Остаточно встановити групу симетрії і координати базисних атомів складних структур можливо лише за допомогою складного аналізу і трудомісткої математичної обробки значень інтенсивностей всіх дифракційних віддзеркалень від даного кристала. Кінцева мета такої обробки полягає в обчисленні за експериментальними даними значень електронної щільності r( х, в, z ) в будь-якій точці вічка кристала з координатами x , в, z. Періодичність будови кристала дозволяє записати електронну щільність в нім через Фур'є ряд :
, (2)
де V — об'єм елементарного вічка, F hkl — коефіцієнти Фур'є, які в Р. с. а. називаються структурними амплітудами, i = . Кожна структурна амплітуда характеризується трьома цілими числами hkl і пов'язана з тим дифракційним віддзеркаленням, яке визначається умовами (1). Призначення підсумовування (2) — математично зібрати дифракційні рентгенівські віддзеркалення, щоб отримати зображення атомної структури. Виробляти таким чином синтез зображення в Р. с. а. доводиться через відсутність в природі лінз для рентгенівського випромінювання (у оптиці видимого світла для цього служить збираюча лінза).
Дифракційне віддзеркалення — хвилевий процес. Він характеризується амплітудою, рівною ½ F hk l ½, і фазою a hkl (зрушенням фази відбитої хвилі по відношенню до падаючої), через яку виражається структурна амплітуда: F hk l =½ F hkl ½(cosa hkl + i sina hkl ) . Дифракційний експеримент дозволяє вимірювати лише інтенсивності віддзеркалень, пропорційні ½ F hk l ½ 2 , але не їх фази. Визначення фаз складає основну проблему розшифровки структури кристала. Визначення фаз структурних амплітуд в принциповому відношенні однаково як для кристалів, що складаються з атомів, так і для кристалів, що складаються з молекул. Визначивши координати атомів в молекулярному кристалічній речовині, можна виділити складові його молекули і встановити їх розмір і форму.
Легко вирішується завдання, зворотне структурній розшифровці: обчислення по відомій атомній структурі структурних амплітуд, а по ним — інтенсивностей дифракційних віддзеркалень. Метод проб і помилок, історично перший метод розшифровки структур, полягає в зіставленні експериментально отриманих ½ F hk l ½ експ , з обчисленими на основі пробної моделі значеннями ½ F hk l ½ вич . Залежно від величини чинника расходімості
пробна модель приймається або відкидається. У 30-х рр. були розроблені для кристалічних структур формальніші методи, але для некристалічних об'єктів метод проб і помилок як і раніше є практично єдиним засобом інтерпретації дифракційної картини.
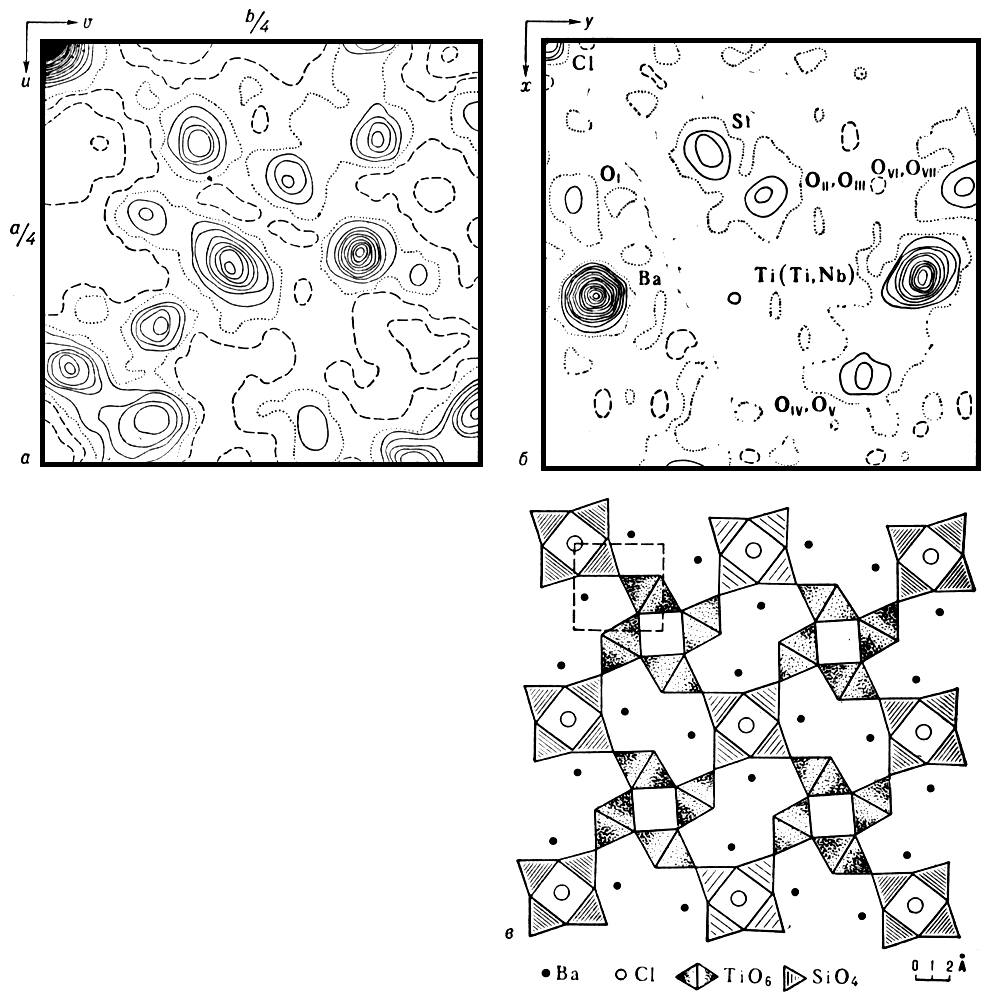
Принципово нову дорогу до розшифровки атомних структур монокристалів відкрило вживання т.з. функцій Патерсона (функцій міжатомних векторів). Для побудови функції Патерсона деякої структури, що складається з N атомів, перенесемо її паралельно самій собі так, щоб у фіксований початок координат попав спочатку перший атом. Вектори від початки координат до всіх атомів структури (включаючи вектор нульової довжини до першого атома) вкажуть положення N максимумів функції міжатомних векторів, сукупність яких називається зображенням структури в атомі 1. Додамо до них ще N максимумів, положення яких вкаже N векторів від другого атома, поміщеного при паралельному перенесенні структури в той же початок координат. Виконавши цю процедуру зі всіма N атомами ( мал. 3 ), ми отримаємо N 2 векторів. Функція, що описує їх положення, і є функція Патерсона.
Для функції Патерсона Р ( u, u, w ) ( u, u, w — координати крапок в просторі міжатомних векторів) можна отримати вираження:
,
з якого виходить, що вона визначається модулями структурних амплітуд, не залежить від їх фаз і, отже, може бути обчислена безпосередньо за даними дифракційного експерименту. Трудність інтерпретації функції Р ( u, u, w ) полягає в необхідності знаходження координат N атомів з N 2 єє максимумів, багато хто з яких зливається із-за перекриттів, що виникають при побудові функції міжатомних векторів. Найбільш простий для розшифровки Р ( u, u, w ) випадок, коли в структурі міститься один важкий атом і декілька легенів. Зображення такої структури у важкому атомі значно відрізнятиметься від ін. її зображень. Серед різних методик, що дозволяють визначити модель досліджуваної структури по функції Патерсона, найбільш ефективними виявилися так звані суперпозиційні методи, які дозволили формалізувати її аналіз і виконувати його на ЕОМ(електронна обчислювальна машина).
Методи функції Патерсона стикаються з серйозними труднощами при дослідженні структур кристалів, що складаються з однакових плі близьких по атомному номеру атомів. В цьому випадку ефективнішими виявилися Так звані прямі методи визначення фаз структурних амплітуд. Враховуючи той факт, що значення електронної щільності в кристалі завжди позитивно (або дорівнює нулю), можна отримати велике число нерівностей, яким підкоряються коефіцієнти Фур'є (структурні амплітуди) функції r( x , в, z ). Методами нерівностей можна порівняно просто аналізувати структури, що містять до 20—40 атомів в елементарному вічку кристала. Для складніших структур застосовуються методи, засновані на імовірнісному підході до проблеми: структурні амплітуди і їх фази розглядаються як випадкові величини; з фізичних вистав виводяться функції розподілу цих випадкових величин, які дають можливість оцінити з врахуванням експериментальних значень модулів структурних амплітуд найбільш вірогідні значення фаз. Ці методи також реалізовані на ЕОМ(електронна обчислювальна машина) і дозволяють розшифрувати структури, що містять 100—200 і більш за атоми в елементарному вічку кристала.
Отже, якщо фази структурних амплітуд встановлені, то по (2) може бути обчислене розподіл електронної щільності в кристалі, максимуми цього розподіли відповідають положенню атомів в структурі ( мал. 4 ). Завершальне уточнення координат атомів проводиться на ЕОМ(електронна обчислювальна машина) найменших квадратів методом і залежно від якості експерименту і складності структури дозволяє отримати їх з точністю до тисячних доль (за допомогою сучасного дифракційного експерименту можна обчислювати також кількісні характеристики теплових коливань атомів в кристалі з врахуванням анізотропії цих коливань). Р. с. а. дає можливість встановити і тонші характеристики атомних структур, наприклад розподіл валентних електронів в кристалі. Проте це складне завдання вирішене доки лише для простих структур. Вельми перспективно для цієї мети поєднання нейтронографічеських і рентгенографічних досліджень: нейтронографічеськие дані про координати ядер атомів зіставляють з розподілом в просторі електронної хмари, отриманим за допомогою Р. с. а. Для вирішення багатьох фізичних і хімічних завдань спільно використовують рентгеноструктурниє дослідження і резонансні методи.
Вершина досягнень Р. с. а. — розшифровка тривимірної структури білків, нуклеїнових кислот і інших макромолекул. Білки в природних умовах, як правило, кристалів не утворюють. Щоб добитися регулярного розташування білкових молекул, білки кристалізують і потім досліджують їх структуру. Фази структурних амплітуд білкових кристалів можна визначити лише в результаті спільних зусиль рентгенографов і біохіміків. Для вирішення цієї проблеми необхідно отримати і досліджувати кристали самого білка, а також його похідних з включенням важких атомів, причому координати атомів у всіх цих структурах повинні збігатися.
Про багаточисельні вживання методів Р. с. а. для дослідження різних порушень структури твердих тіл під впливом всіляких дій див.(дивися) в ст. Рентгенографія матеріалів.
Літ.: Белов Н. Ст, Структурна кристалографія, М., 1951; Жданов Р. С., Основи рентгеноструктурного аналізу, М. — Л., 1940; Джеймс Р., Оптичні принципи дифракція рентгенівських променів, пер.(переведення) з англ.(англійський), М., 1950; Бокий Р. Би., Порай-Кошиц М. А., Рентгеноструктурний аналіз, М., 1964; Порай-Кошиц М. А., Практичний курс рентгеноструктурного аналізу, М., 1960: Китайгородський А. І., Теорія структурного аналізу, М., 1957; Ліпеон Р., Кокрен Ст, Визначення структури кристалів, пер.(переведення) з англ.(англійський), М., 1961; Вайнштейн Би. До., Структурна електронографія, М., 1956; Бекон Дж., Дифракція нейтронів пер.(переведення) з англ.(англійський), М., 1957; Бюргер М., Структура кристалів і векторний простір, пер.(переведення) з англ.(англійський), М., 1961; Гинье А., Рентгенографія кристалів, пер.(переведення) з франц.(французький), М., 1961; Woolfson М. М., An introduction to X-ray crystallography, Camb., 1970: Ramachandran G. N., Srinivasan R., Fourier methode in crystallography, N. Y., 1970; Crystallographic computing, ed. F. R. Ahmed, Cph., 1970; Stout G. H., Jensen L. H., X-ray structure determination, N. Y. — L., [1968].