Спектральный анализ, физический метод качественного и количественного определения атомного и молекулярного состава вещества, основанный на исследовании его спектров. Физическая основа С. а.— спектроскопия атомов и молекул, его классифицируют по целям анализа и типам спектров (см. Спектры оптические). Атомный С. а. (АСА) определяет элементный состав образца по атомным (ионным) спектрам испускания и поглощения, молекулярный С. а. (МСА) — молекулярный состав веществ по молекулярным спектрам поглощения, люминесценции и комбинационного рассеяния света.
Эмиссионный С. а. производят по спектрам испускания атомов, ионов и молекул, возбуждённым различными источниками электромагнитного излучения в диапазоне от g-излучения до микроволнового. Абсорбционный С. а. осуществляют по спектрам поглощения электромагнитного излучения анализируемыми объектами (атомами, молекулами, ионами вещества, находящегося в различных агрегатных состояниях).
Историческая справка. В основе АСА лежит индивидуальность спектров испускания и поглощения химических элементов, установленная впервые Г. Р. Кирхгофоми Р. Бунзеном (1859—61). В 1861 Кирхгоф доказал на основе этого открытия присутствие в хромосфере Солнца ряда элементов, положив начало астрофизике. В 1861—1923 с помощью АСА было открыто 25 элементов. В 1932 спектральным методом был открыт дейтерий.
Высокая чувствительность и возможность определения многих элементов в пробах малой массы сделали АСА эффективным методом качественного анализа элементного состава объектов. В 1926 нем.(немецкий) физик В. Герлах положил начало количественному С. а. Для развития С. а. и внедрения его на промышленных предприятиях СССР большую роль сыграли Г. С. Ландсберг, С. Л. Мандельштам, А. К. Русанов (Москва), А. Н. Филиппов, В. К. Прокофьев (Ленинград) и др.
Атомный спектральный анализ (АСА)
Эмиссионный АСА состоит из следующих основных процессов:
1) отбор представительной пробы, отражающей средний состав анализируемого материала или местное распределение определяемых элементов в материале;
2) введение пробы в источник излучения, в котором происходят испарение твёрдых и жидких проб, диссоциация соединений и возбуждение атомов и ионов;
3) преобразование их свечения в спектр и его регистрация (либо визуальное наблюдение) с помощью спектрального прибора;
4) расшифровка полученных спектров с помощью таблиц и атласов спектральных линий элементов.
На этой стадии заканчивается качественный АСА. Наиболее результативно использование чувствительных (т. н. «последних») линий, сохраняющихся в спектре при минимальной концентрации определяемого элемента. Спектрограммы просматривают на измерительных микроскопах, компараторах, спектропроекторах. Для качественного анализа достаточно установить наличие или отсутствие аналитических линий определяемых элементов. По яркости линий при визуальном просмотре можно дать грубую оценку содержания тех или иных элементов в пробе.
Количественный АСА осуществляют сравнением интенсивностей двух спектральных линий в спектре пробы, одна из которых принадлежит определяемому элементу, а другая (линия сравнения) — основному элементу пробы, концентрация которого известна, или специально вводимому в известной концентрации элементу («внутреннему стандарту»).
В основе количественного АСА лежит соотношение, связывающее концентрацию с определяемого элемента с отношением интенсивностей линии определяемой примеси (I1) и линии сравнения (I2):
I1/I2 = acb
(постоянные а и b определяются опытным путём), или
lg(I1/I2) = b lgс + lga.
С помощью стандартных образцов (не менее 3) можно построить график зависимости lg(I1/I2.) от lg с (градуировочный график, рис. 1) и определить по нему а и b. Значения I1 и I2 можно получать непосредственно путём фото-электрической регистрации или путём фотометрирования (измерения плотности почернения) линии определяемой примеси и линии сравнения при фоторегистрации. Фотометрирование производят на микрофотометрах.
Для возбуждения спектра в АСА используют различные источники света и соответственно различные способы введения в них образцов. Выбор источника зависит от конкретных условий анализа определённых объектов. Тип источника и способ введения пробы составляют главное содержание частных методик АСА.
Первым искусственным источником света в АСА было пламя газовой горелки — источник весьма удобный для быстрого и точного определения многих элементов. Температура пламён горючих газов не высока (от 2100 К для смеси водород — воздух до 4500 К для редко используемой смеси кислород — циан). С помощью фотометрии пламени определяют около 70 элементов по их аналитическим линиям, а также по молекулярным полосам соединений, образующихся в пламёнах.
В эмиссионном АСА широко используют электрические источники света. В электрической дуге постоянного тока между специально очищенными угольными электродами различной формы, в каналы которых помещают исследуемое вещество в измельченном состоянии, можно производить одновременное определение десятков элементов. Она обеспечивает относительно высокую температуру нагрева электродов и благоприятные условия возбуждения атомов пробы в дуговой плазме, однако точность этого метода невысока из-за нестабильности разряда. Повышая напряжение до 300—400 в или переходя к высоковольтной дуге (3000—4000 в), можно увеличить точность анализа.
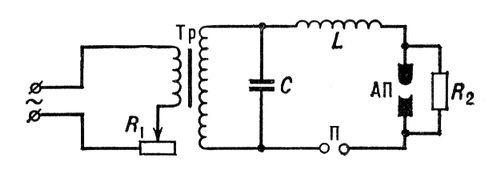
Более стабильные условия возбуждения создаёт дуга переменного тока. В современных генераторах дуги переменного тока (см., напр., рис.(рисунок) 2) можно получить различные режимы возбуждения: низковольтную искру, высокочастотную искру, дугу переменного тока, импульсный разряд и т. д. Такие источники света с различными режимами используют при определении металлов и трудновозбудимых элементов (углерод, галогены, газы, содержащиеся в металлах, и т. д.). Высоковольтная конденсированная искра (рис. 3) служит главным образом источником света при анализе металлов. Стабильность искрового разряда позволяет получать высокую воспроизводимость анализа, однако сложные процессы, происходящие на поверхностях анализируемых электродов, приводят к изменениям состава плазмы разряда. Чтобы устранить это явление, приходится производить предварительный обжиг проб и нормировать форму и размеры проб и стандартных образцов.
В АСА перспективно применение стабилизированных форм электрического разряда типа плазмотронов различных конструкций, высокочастотного индукционного разряда, СВЧ(сверхвысокие частоты)-разряда, создаваемого магнетронными генераторами, высокочастотного факельного разряда. С помощью различных приёмов введения анализируемых веществ в плазму этих типов разряда (продувка порошков, распыление растворов и т. д.) значительно повышена относительная точность анализа (до 0,5—3% ), в том числе и компонентов сложных проб, содержание которых составляет десятки %. В некоторых важных случаях анализа чистых веществ применение этих типов раз ряда снижает пределы определения примесей на 1—2 порядка (до 10-5—10-6 % ).
Для анализа чистых веществ, радиоактивных материалов, смесей газов, изотопного анализа, спектрально-изотопного определения газов в металлах и твёрдых веществах и т. д. весьма перспективным оказалось использование разряда в полом катоде и безэлектродных ВЧ(высокая частота)-и СВЧ(сверхвысокие частоты)-разрядов. В АСА в качестве источников возбуждения применяются также лазеры (см. Спектроскопия лазерная).
Атомно-абсорбционный С. а. (ААА) и атомно-флуоресцентный С. а. (АФА). В этих методах пробу превращают в пар в атомизаторе (пламени, графитовой трубке, плазме стабилизированного ВЧ(высокая частота)-или СВЧ(сверхвысокие частоты)-разряда). В ААА свет от источника дискретного излучения, проходя через этот пар, ослабляется и по степени ослабления интенсивностей линий определяемого элемента судят о концентрации его в пробе. ААА проводят на специальных спектрофотометрах. Методика проведения ААА по сравнению с др. методами значительно проще, для него характерна высокая точность определения не только малых, но и больших концентраций элементов в пробах. ААА с успехом заменяет трудоёмкие и длительные химические методы анализа, не уступая им в точности .
В АФА атомные пары пробы облучают светом источника резонансного излучения и регистрируют флуоресценцию определяемого элемента. Для некоторых элементов (Zn, Cd, Hg и др.) относительные пределы их обнаружения этим методом весьма малы (~10-5—106 %).
АСА позволяет проводить измерения изотопного состава. Некоторые элементы имеют спектральные линии с хорошо разрешенной структурой (например, Н, Не, U). Изотопный состав этих элементов можно измерять на обычных спектральных приборах с помощью источников света, дающих тонкие спектральные линии (полый катод, безэлектродные ВЧ(высокая частота)-и СВЧ(сверхвысокие частоты)-лампы). Для проведения изотопного спектрального анализа большинства элементов требуются приборы высокой разрешающей способности (например, эталон Фабри — Перо). Изотопный спектральный анализ можно также проводить по электронно-колебательным спектрам молекул, измеряя изотопные сдвиги полос, достигающие в ряде случаев значительной величины.
Экспрессные методы АСА широко применяются в промышленности, сельском хозяйстве, геологии и многих др. областях народного хозяйства и науки. Значительную роль АСА играет в атомной технике, производстве чистых полупроводниковых материалов, сверхпроводников и т. д. Методами АСА выполняется более 3/4 всех анализов в металлургии. С помощью квантометров проводят оперативный (в течение 2—3 мин) контроль в ходе плавки в мартеновском и конвертерном производствах. В геологии и геологической разведке для оценки месторождений производят около 8 млн. анализов в год. АСА применяется для охраны окружающей среды и анализа почв, в криминалистике и медицине, геологии морского дна и исследовании состава верхних слоев атмосферы, при разделении изотопов и определении возраста и состава геологических и археологических объектов и т. д.
Лит.: Заидель А. Н., Основы спектрального анализа, М., 1965; Методы спектрального анализа, М,, 1962; Эмиссионный спектральный анализ атомных материалов, Л. — М., 1960; Русанов А. К., Основы количественного спектрального анализа руд и минералов. М., 1971; Спектральный анализ чистых веществ, под ред. X. И. Зильберштейна, [Л.], 1971; Львов Б. В., Атомно-абсорбционный спектральный анализ, М., 1966; Петров А. А., Спектрально-изотопный метод исследования материалов, Л., 1974; Тарасевич Н. И.. Семененко К. А., Хлыстова А. Д., Методы спектрального и химико-спектрального анализа, М., 1973: Прокофьев В. К., Фотографические методы количественного спектрального анализа металлов и сплавов, ч. 1—2, М. — Л., 1951; Менке Г., Менке Л., Введение в лазерный эмиссионный микроспектральный анализ, пер.(перевод) с нем.(немецкий), М., 1968; Королев Н. В., Рюхин В. В., Горбунов С. А., Эмиссионный спектральный микроанализ, Л., 1971; Таблицы спектральных линий, 3 изд., М., 1969; Стриганов A. P., Свентицкий Н. С., Таблицы спектральных линий нейтральных и ионизованных атомов, М., 1966.
Л. В. Липис.
Молекулярный спектральный анализ (МСА)
В основе МСЛ лежит качественное и количественное сравнение измеренного спектра исследуемого образца со спектрами индивидуальных веществ. Соответственно различают качественный и количественный МСА. В МСА используют различные виды молекулярных спектров, вращательные [спектры в микроволновой и длинноволновой инфракрасной (ИК) областях], колебательные и колебательно-вращательные [спектры поглощения и испускания в средней ИК-области, спектры комбинационного рассеяния света (КРС), спектры ИК-флуоресценции], электронные, электронно-колебательные и электронно-колебательно-вращательные [спектры поглощения и пропускания в видимой и ультрафиолетовой (УФ) областях, спектры флуоресценции]. МСА позволяет проводить анализ малых количеств (в некоторых случаях доли мкг и менее) веществ, находящихся в различных агрегатных состояниях.
Основные факторы, определяющие возможности методов МСА:
1) информативность метода. Условно выражается числом спектрально разрешаемых линий или полос в определённом интервале длин волн или частот исследуемого диапазона (для микроволнового диапазона оно ~ 105, для средней ИК-области в спектрах твёрдых и жидких веществ ~ 103);
2) количество измеренных спектров индивидуальных соединений;
3) существование общих закономерностей между спектром вещества и его молекулярным строением;
4) чувствительность и избирательность метода;
5) универсальность метода;
6) простота и доступность измерений спектров.
Качественный МСА устанавливает молекулярный состав исследуемого образца. Спектр молекулы является его однозначной характеристикой. Наиболее специфичны спектры веществ в газообразном состоянии с разрешенной вращательной структурой, которые исследуют с помощью спектральных приборов высокой разрешающей способности. Наиболее широко используют спектры ИК-поглощения и КРС веществ в жидком и твёрдом состояниях, а также спектры поглощения в видимой и УФ-областях. Широкому внедрению метода КРС способствовало применение для их возбуждения лазерного излучения.
Для повышения эффективности МСА в некоторых случаях измерение спектров комбинируют с др. методами идентификации веществ. Так, всё большее распространение получает сочетание хроматографического разделения смесей веществ с измерением ИК-спектров поглощения выделенных компонент.
К качественному МСА относится также т. н. структурный молекулярный анализ. Установлено, что молекулы, имеющие одинаковые структурные элементы, обнаруживают в спектрах поглощения и испускания общие черты. Наиболее ярко это проявляется в колебательных спектрах. Так, наличие сульфгидрильной группы (—SH) в структуре молекулы влечёт за собой появление в спектре полосы в интервале 2565—2575 см-1, нитрильная группа (—CN) характеризуется полосой 2200—2300 cм-1 и т. д. Присутствие таких характеристических полоса колебательных спектрах веществ с общими структурными элементами объясняется характеристичностью частоты и формы многих молекулярных колебаний. Подобные особенности колебательных (и в меньшей степени электронных) спектров во многих случаях позволяют определять структурный тип вещества.
Качественный анализ существенно упрощает и ускоряет применение ЭВМ(электронная вычислительная машина). В принципе его можно полностью автоматизировать, вводя показания спектральных приборов непосредственно в ЭВМ(электронная вычислительная машина). В её памяти должны быть заложены спектральные характеристические признаки многих веществ, на основании которых машина произведёт анализ исследуемого вещества.
Количественный МСА по спектрам поглощения основан на Бугера — Ламберта — Бера законе, устанавливающем связь между интенсивностями падающего и прошедшего через вещество I света от толщины поглощающего слоя I и концентрации вещества с:
I(l)=I0e-ccl
Коэффициент c является характеристикой поглощающей способности определяемого компонента для данной частоты излучения. Важное условие проведения количественного МСА — независимость c от концентрации вещества и постоянство c в измеряемом интервале частот, определяемом шириной щели спектрофотометра. МСА по спектрам поглощения проводят преимущественно для жидкостей и растворов, для газов он значительно усложняется.
В практическом МСА обычно измеряют т. н. оптическую плотность:
D = In (/о//) = cсl.
Если смесь состоит из n веществ, не реагирующих друг с другом, то оптическая плотность смеси на частоте n аддитивна: . Это позволяет проводить полный или частичный анализ многокомпонентных смесей. Задача в этом случае сводится к измерению значений оптической плотности в m точках спектра смеси (m ³ n) и решению получаемой системы уравнений:
Для количественного МСА обычно пользуются спектрофотометрами, позволяющими производить измерение /(n) в сравнительно широком интервале n . Если полоса поглощения исследуемого вещества достаточно изолирована и свободна от наложения полос др. компонент смеси, исследуемый спектральный участок можно выделить, например, при помощи интерференционного светофильтра. На его основе конструируют специализированные анализаторы, широко используемые в промышленности.
При количественном МСА по спектрам КРС чаще всего интенсивность линии определяемого компонента смеси сравнивают с интенсивностью некоторой линии стандартного вещества, измеренной в тех же условиях (метод «внешнего стандарта»). В др. случаях стандартное вещество добавляют к исследуемому в определённом количестве (метод «внутреннего стандарта» ).
Среди др. методов качественного и количественного МСА наибольшей чувствительностью обладает флуоресцентный анализ, однако в обычных условиях он уступает методам колебательной спектроскопии в универсальности и избирательности. Количественный МСА по спектрам флуоресценции основан на сравнении свечения раствора исследуемого образца со свечением ряда эталонных растворов близкой концентрации.
Особое значение имеет МСА с применением техники замороженных растворов в специальных растворителях, например парафинах (см. Шпольского эффект). Спектры веществ в таких растворах (спектры Шпольского) обладают ярко выраженной индивидуальностью, они резко различны для близких по строению и даже изомерных молекул. Это позволяет идентифицировать вещества, которые по спектрам их флуоресценции в обычных условиях установить не удаётся. Например, метод Шпольского даёт возможность осуществлять качественный и количественный анализ сложных смесей, содержащих ароматические углеводороды. Качественный анализ в этом случае производят по спектрам люминесценции и поглощения, количественный — по спектрам люминесценции методами «внутреннего» и «внешнего» стандартов. Благодаря исключительно малой ширине спектральных линий в спектрах Шпольского в этом методе удаётся достигнуть пороговой чувствительности обнаружения некоторых многоатомных ароматических соединений (~ 10~11г/см3).
Лит.: Чулановский В. М., Введение в молекулярный спектральный анализ, М. — Л., 1951; Беллами Л., Инфракрасные спектры сложных молекул, пер.(перевод) с англ.(английский), М., 1963; Применение спектроскопии в химии, пер.(перевод) с англ.(английский), М., 1959; Определение индивидуального углеводородного состава бензинов прямой гонки комбинированным методом, М., 1959; Юденфренд С., Флуоресцентный анализ в биологии и медицине, пер.(перевод) с англ.(английский), М., 1965.